Abstract
Psoriasis affects 60 million people globally. It is an inflammatory skin condition that can significantly impact quality of life. Furthermore, comorbidities including psoriatic arthritis and cardiovascular disease are common, indicating wider immune system involvement. In recent years, the IL-23/Type 17 T cells (T17) axis of inflammation has been shown to play a key role in psoriasis pathogenesis, leading to the development and licensing of biologics that target IL-23- and IL-17-mediated inflammatory pathways. These advanced medications effectively disrupt the inflammatory cycle, resulting in sustained improvements in clinical disease. This article reviews newly available data that provide the latest insights into the molecular interactions and mode of action of IL-23 antagonists. It outlines evidence to demonstrate that IL-23 blockade with risankizumab selectively inhibits pathogenic T17 cell subsets in psoriasis skin lesions, while non-pathogenic or regulatory T17 subsets remain intact, as well as the potential clinical benefits. It also evaluates the alternative binding to CD64 and concludes that it is unlikely to affect the clinical efficacy or safety of licensed IL-23 antibodies. These new data provide clinically relevant insights into the durable efficacy and safety of anti-IL-23 biologics.
INTRODUCTION
Psoriasis is an inflammatory skin condition that affects more than 60 million people worldwide.1 Several subtypes exist, but the most prevalent is plaque psoriasis, also known as psoriasis vulgaris.2 The condition manifests on the skin as erythematous, scaly patches of varying size that can coalesce and appear on any part of the body. Importantly, inflammation is not limited to skin and can affect joints, causing psoriatic arthritis.2,3 Comorbidities are also common, including cardiovascular disease, metabolic syndrome, and inflammatory bowel disease.4 The condition has a significant negative impact on quality of life, causing disrupted sleep, low self-esteem, and social isolation, and can lead to cumulative life course impairment over a patient’s lifetime.5
Characteristic psoriasis skin lesions harbour hyperplastic keratinocytes in the epidermis and a variety of infiltrating leukocytes, including dendritic cells and T cells, in the dermis.6,7 This infiltration is triggered when dermal dendritic cells are activated to secrete inflammatory mediators, including the cytokine IL-23, which drives the differentiation of a subset of T cells, called Type 17 T cells (T17).3 T17 cells produce the inflammatory cytokines IL-17 and IL-22 that drive overproduction of keratinocytes, resulting in plaque formation.6
Recent advances in the understanding of the IL-23/T17 axis of inflammation in psoriasis have led to the development and licensing of effective targeted treatments for moderate-to-severe psoriasis and psoriatic arthritis, including biologics such as IL-23 and IL-17 inhibitors that disrupt the cycle of psoriatic inflammation.8,9
This article focuses on recently published data that shed new light on the mode of action of IL-23 inhibitors and their effectiveness as treatments for psoriasis, demonstrating their targeted effect in reducing pathogenic T cell subsets in psoriatic lesions.
CELL-MEDIATED INFLAMMATION IN PSORIASIS
In psoriasis, inflammation can be considered to occur in two phases.2 In the initial phase, activated dendritic cells in the skin produce IL-23 in the form of heterodimers (consisting of a p19 and a p40 subunit), which bind to IL-23 receptors expressed on the surface of cells, including T17 cells.10 Signalling via the IL-23 receptor stimulates T17 cells to produce cytokines, including IL-17 and IL-22. While IL-23 acts as a regulatory cytokine in the early stages of the inflammatory pathway in psoriasis,11 effector cytokines, including IL-17, produced by T17 cells, maintain inflammation,12 stimulating keratinocytes to proliferate and secrete other inflammatory mediators, resulting in plaque formation and maintenance (Figure 1).3,10 This IL-23/T17 immune axis is now known to be instrumental in psoriasis and in several other autoimmune conditions, including inflammatory bowel disease,12 and raised levels of IL-23 can be detected in psoriatic skin lesions.10
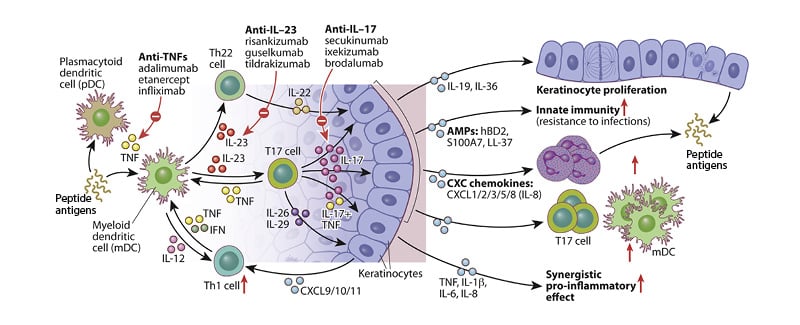
Figure 1: Schematic showing IL-23/T17-mediated effects on epidermal keratinocytes in psoriatic skin.
Adapted from Hawkes JE et al.13
TARGETING THE IL-23/T17 AXIS OF INFLAMMATION IN PSORIASIS
Several novel biologics targeting IL-23 or IL-17 have been developed as therapeutic interventions for moderate-to-severe psoriasis.12 For example, in global Phase III trials, over 70% of participants treated with the IL-23 inhibitor risankizumab (150 mg) experienced substantial skin clearance and clinical improvement in the extent and severity of plaque psoriasis after 4 months.14 Most patients maintained clear (Psoriasis Area and Severity Index [PASI] 100), or almost clear (PASI 90), skin for 5 years, and this was associated with significant improvements in quality of life.15
Blocking the activity of IL-23 with a neutralising antibody directly reduces IL-17–induced inflammation mediated via T17 cells (Figure 2). The antibody binds to IL-23 and blocks its interaction with the IL-23 receptor (IL-23R) found on the surface of T17 cells and some other cell types, resulting in highly potent neutralisation of IL-23 bioactivity and a reduction in the production of IL-17.6 Although IL-23 blockade reduces IL-17 production, it does not completely ablate it, and T17 cells can produce some IL-17 independently of signalling via the IL-23R.6
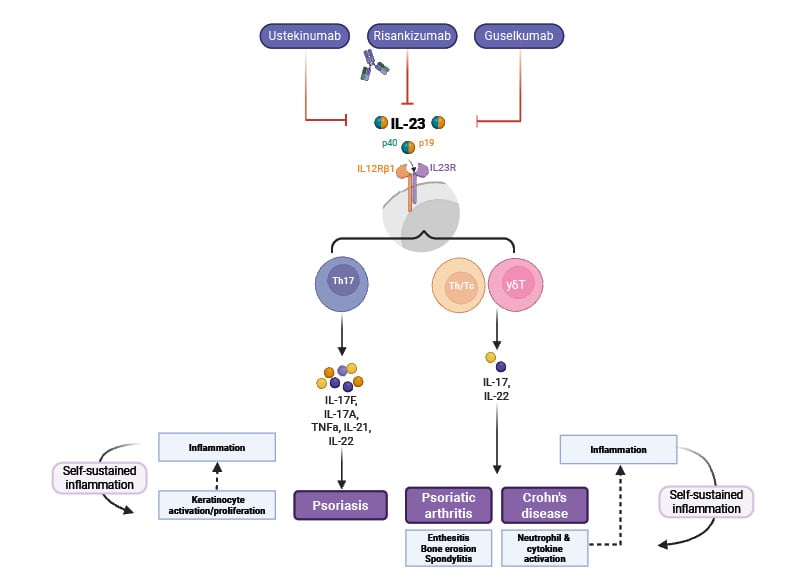
Figure 2: Overview of mode of action of IL-23 inhibitors.
Adapted from Pang Y et al.10
Created using BioRender.com
There is growing evidence that different types of T17 cells exist, as demonstrated using murine models,16 including non-pathogenic (homeostatic) and pathogenic T17 cells.11 Recent in vitro studies using human skin samples,6 and in animal models,11 show that the activation of the IL-23R is responsible for pathogenic effector T17 cell responses, which include the production of other inflammatory cytokines, including IL-22, GM-CSF, and IFN-γ.6,11 While non-pathogenic T17 cells regulate barrier functions and control microbial invasion at mucosal surfaces, pathogenic T17 cells induce tissue inflammation and autoimmunity.11 Recent research in vitro using a mouse model has shown that IL-23R expression differentiates pathogenic from non-pathogenic T17 cells, with pathogenic cells demonstrating high levels of IL-23R expression.16
SELECTIVE DOWNREGULATION OF PATHOGENIC T17 CELL SUBSETS BY RISANKIZUMAB
Disrupting the IL-23/IL-17 inflammatory axis by blocking either lL-23 or IL-17 has been shown to control psoriasis effectively.17-20 However, there appear to be differences between IL-23 and IL-17 blockade, as well as among IL-23 inhibitors.
In head-to-head clinical trials, it was shown that IL-23 blockade with risankizumab or guselkumab was superior to secukinumab (IL-17 blockade) after 1 year.17,18 In addition, IL-23 plays a pivotal role in the maintenance and function of tissue-resident memory T cells in various tissues, including the skin.21 In the context of psoriasis, IL-23 is essential for the persistence of skin-resident memory Th17 cells, which are implicated in the chronicity and recurrence of the disease.11 Clinical evidence showed that patients who were withdrawn from treatment with IL-17 inhibitors lost their response to treatment faster than patients on anti-IL-23 treatment.22 Trials have shown that the median time to loss of PASI 90 response (clear or almost clear skin) of 210 days for risankizumab compares with 106 days for guselkumab and 126 days for tildrakizumab, although these were not head-to-head studies.23-25 Whereas, time to relapse following secukinumab treatment was 168–196 days (300 mg dose).26,27
To further investigate the complexities of the T17 subsets involved in psoriasis pathogenesis and differences in the mode of action of IL-23 and IL-17 inhibitors, Kim et al.7 explored, at a single cell level, the genomic profiles of T17 cell subsets in psoriasis skin before and after treatment with risankizumab or secukinumab.17,18 They found that, although expression of IL-23R transcripts was reduced in skin lesions following IL-23 inhibition, transcripts for IL-17 and IL-23R were still detectable following treatment. These data demonstrate that systemic IL-23 inhibition substantially downregulates but does not eradicate the expression of IL-23/T17 pathway genes in the skin, even at a point where systemic IL-23 inhibition has maximal impact on reducing skin lesions.7
Prior to the study by Kim et al.,7 pathogenic versus non-pathogenic subsets of T17 cells were not known. The presence of IL-23/T17 cell transcriptomes in the clinically improved psoriasis skin after systemic IL-23 inhibition led Kim et al.7 to investigate whether IL-23 inhibition selectively downregulates pathogenic T17 subsets, leaving non-pathogenic T17 subsets present in the post-treatment psoriasis skin.7 By comparing the single-cell transcriptome profiles of different T17 subsets within psoriasis skin samples, the investigators found that a T17 subset that expressed IL-23R transcripts and IL-17A and IFN-γ transcripts was present in lesions before treatment, but was eradicated after IL-23 inhibition. Another IL-23R-expressing pathogenic T17 subset (IL17F+, IL10-subset) was also selectively downregulated by IL-23 blockade. At the same time, non-pathogenic T cell subsets that did not express IL-23 transcripts were retained following the anti-IL-23 treatment (Figure 3A). The investigators also found that, as the percentages of pathogenic T17 subsets decreased, the percentage of a non-pathogenic T17 subset increased (Figure 3B).7 The authors conclude that, considering that IL-23 inhibition is not associated with an increased risk of candidiasis, unlike IL-17 inhibition, which is associated with an increased risk of candidiasis,28 the IL-17A+ IL-17F+ T17 subset may represent the non-pathogenic skin-resident memory Th17 cells preserved after IL-23 inhibition because of its lack of IL-23R expression, defending skin against external surface microbes after treatment.
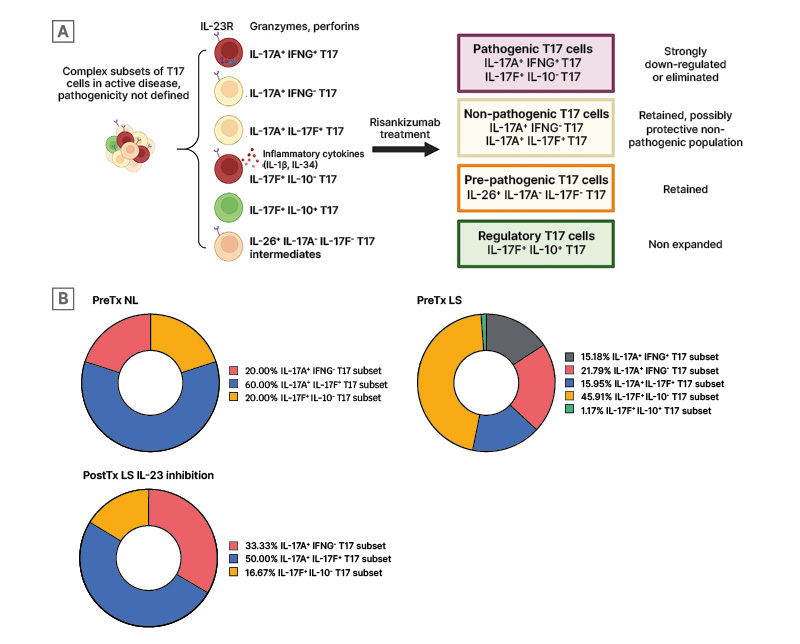
Figure 3: Selective modulation of pathogenic T17 subsets by risankizumab.
A) Analysis of single T cell phenotypes in response to IL-23 inhibition demonstrates selective downregulation of pathogenic T17 subsets, while pre-pathogenic T17 cells and regulatory T17 cells are retained. B) Proportional changes of T17 subsets between Pre-Tx NL and LS, and Post-tx LS IL-23 inhibition.
Adapted from Kim J et al.7
LS: lesional skin; NL: non-lesional; Post-Tx: post-treatment; Pre-Tx: pre-treatment; T17: Type 17 T cells.
STRUCTURAL DIFFERENCES IN IL-23 INHIBITORS
Four monoclonal antibodies that neutralise IL-23 activity are licensed as treatments for psoriasis (risankizumab, guselkumab, ustekinumab, and tildrakizumab), and all have demonstrated efficacy in reducing psoriatic skin lesions.19 Three of the monoclonal antibodies (risankizumab, guselkumab, and tildrakizumab) bind distinct epitopes on the p19 subunit of the IL-23 heterodimer.6 Risankizumab and guselkumab inhibit the interaction of IL-23 with its receptor, IL-23Rα, through competitive inhibition of IL-23. In contrast, tildrakizumab-bound IL-23 is capable of binding to IL-23Rα, but at a 200-fold weaker affinity.6
Risankizumab is a human IgG1 antibody that was engineered with two mutations in the Fc region (L234A, L235A mutations, known as LALA) that lower its binding to Fc gamma receptors, including CD64 and complement C1q, to avoid any unwanted cellular cytotoxicity.10 LALA mutations have no effect on neonatal FcR binding; thus, risankizumab exhibits linear and time-independent pharmacokinetic characteristics consistent with typical IgG1 monoclonal antibodies,10 and demonstrates high affinity and potency, with the longest half-life among the approved IL-23 monoclonal antibodies, in patients with psoriasis, of 28 days.10
In contrast to risankizumab, the other licensed anti-IL-23 biologics, guselkumab and tildrakizumab, do not have mutations in the Fc region. While in vitro studies show that guselkumab exhibits strong binding to CD64 expressed on cell lines,29 in the presence of plasma, neither guselkumab nor risankizumab bind to CD64 expressed on activated monocytes from healthy human donors.29 Indeed, the much higher concentration of endogenous IgG1 in vivo or in plasma competitively blocks the high affinity Fc receptor, CD64, which prevents binding of the anti-IL-23 biologics to it.29 In addition, in other in vitro cellular assays, when IL-23 was added exogenously, guselkumab and risankizumab demonstrated similar potency for inhibition of IL-23 signalling.30 Although further investigation may provide additional insight, the hypothesis that binding to CD64 is a mechanism of action, based on in vitro data, is unlikely to be clinically relevant in vivo in patients and has not been shown to impact clinical effectiveness or safety.31,32
CONCLUSION
Psoriasis is driven by a complex cascade of cellular events and inflammatory cytokines, including IL-23 and IL-17. Biologics that target the IL-23/T17 axis of inflammation are effective in controlling active disease and demonstrate sustained efficacy even beyond the treatment period. Analysis of T cell subsets from psoriasis lesions now demonstrates that the IL-23 antagonist risankizumab selectively targets and removes pathogenic IL-23R-expressing T17 cells in psoriasis skin lesions. Other T17 subsets in psoriasis skin that are unaffected by IL-23 blockade are likely to mediate protective immunity and may help to prevent skin infections like candidiasis. In addition, insights into the structural binding characteristics of IL-23 inhibitors in plasma support existing clinical data that demonstrate the potential for CD64 binding is unlikely to have any clinical consequences. While these data provide new insights into the mode of action of IL-23 antagonists, it remains unclear whether pathogenic T17 subsets are unique to psoriasis or if they occur in other skin disorders or other target organs. It is, therefore, important to further investigate the role of these different cell subsets.