Meeting Summary
On the 29th May 2025 in Budapest, Hungary, leading experts discussed bradykinin-mediated angioedema (AE-BK). Henriette Farkas, Head of the Hungarian Angioedema Center of Reference and Excellence, Semmelweis University, in Budapest, Hungary; Marc A. Riedl, Clinical Director of the US HAEA Angioedema Center, UC San Diego Health, California, USA; and Danny M. Cohn, Medical Specialist in Vascular Medicine at the Amsterdam University Medical Center, the Netherlands, elucidated the mechanisms underlying the bradykinin (BK) signalling cascade, associated pathways, and the role of BK in both AE-BK and broader inflammatory disorders. The role of BK B2 receptor (B2R) was discussed, as well as the consequences of B2R antagonism, exploring whether this antagonism might have an impact on pathophysiological processes. A key focus was on the unmet needs in AE-BK, notably for on-demand (ODT) and long-term prophylactic (LTP) treatment approaches to address needs in multiple types of AE-BK. For treatments to address individual patient expectations and needs, they should be effective, well-tolerated, and have a low treatment burden regarding portability, handling, and administration. The value of standardised guidelines to aid the classification of recurrent angioedema (AE) was also discussed. Additionally, the value of reliable and accurate biomarker-based testing aiding diagnosis was emphasised. Ongoing research aims to address these gaps by investigating novel biomarker testing approaches and exploring additional therapeutic approaches.
Multifaceted Pathways Lead to Bradykinin Production
BK is a short-lived effector molecule that belongs to the peptide family of kinins and plays a crucial role in inflammation.1,2 It was first discovered in 1949,2 and BK receptors were cloned and characterised in 1994.3,4 In 2004, a B2R antagonist was first evaluated for the treatment of hereditary angioedema (HAE) in the FAST-1 clinical trial.5,6
BK is formed through several biological pathways, including the kallikrein-kinin system (KKS).7 Riedl explained that in physiological conditions, activation of the plasma KKS system and subsequent release of BK is triggered by the contact/coagulation activation system, in response to trauma, injury, or infection.7,8 Contact system activation results in factor XII (FXII) activation, which converts prekallikrein to plasma kallikrein (pKal). pKal cleaves high molecular weight kininogen (HK) to generate BK.7,8 pKal additionally activates FXII, resulting in the amplification of FXII activity and further contact system activation. The activation of both the CAS and KKS systems is physiologically aimed at repairing the injury site.9
BK can also be produced independently of the KKS pathway, through activation of the fibrinolytic pathway, initiated by wound healing and breakdown of fibrin, and results in plasmin generation that can cleave HK, leading to BK generation.10,11
On the other hand, inflammation can also lead to BK generation. Activation of the innate immune system results in degranulation of mast cells and neutrophils to clear pathogens, resulting in release of several proteases that are believed to cleave HK and generate BK.12 Summarising this, Riedl commented: “There are multiple pathways by which BK can be formed, and for the majority, their biological function is related to diseases we are familiar with, all of which are signalled through the common B2R pathway.” Moreover, he highlighted that the production of BK can be influenced by genetics and certain pathogenic mutations, which lead to uncontrolled BK production (Figure 1).
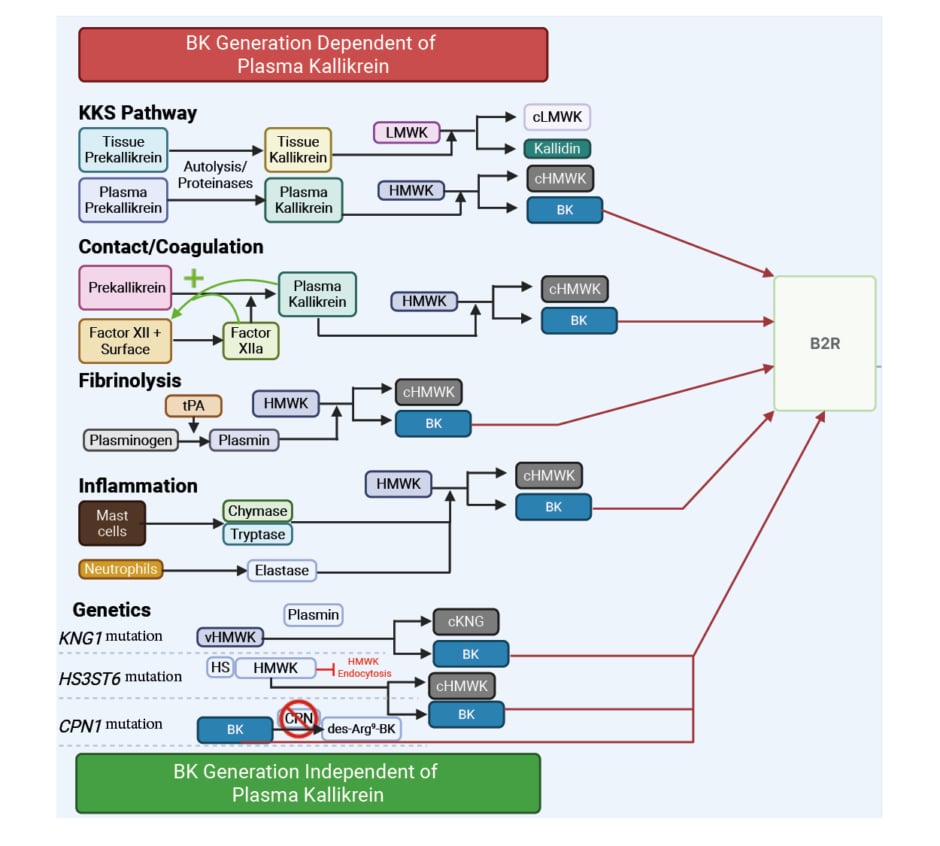
Figure 1: Generation of bradykinin: dependent and independent of plasma kallikrein.
Created in BioRender.com
B2R: bradykinin B2 receptor; BK: bradykinin; cHMWK: cleaved high molecular weight kininogen; cKNG: cleaved kininogen; CPN: carboxypeptidase N; C1INH: C1 inhibitor; des-Arg-BK: bradykinin lacking arginine at position 9; HMWK: high molecular weight kininogen; HS: heparan sulfate; KKS: kallikrein-kinin system; KNG: kininogen; LMWK: low molecular weight kininogen; tPA: tissue-type plasminogen activator; v: variant.
Bradykinin B2 Receptor Antagonism
BK exerts its effects by converging on and activating specific receptors, including the B2R.7,13 B2R, a G protein-coupled receptor (GPCR), is activated through the binding of either BK or, with lower affinity, kallidin.14 Highlighting the clinical relevance of GPCRs such as B2R, Riedl noted: “Approximately one-third of all drugs marketed are targeted at GPCRs, as they are so ubiquitous in disease processes.” BK binds to the B2R through extensive molecular contacts, serving as the primary receptor for BK signalling.7,13,14 Compared to the human BK B1 receptor, BK exhibits a 20,000-fold greater affinity for human B2R.13 Notably, Riedl highlighted that B2R antagonism can modulate BK-mediated effects independently of the pathway leading to BK production.
The B2R is constitutively expressed, and its expression may be elevated in certain pathologies.15,16 Highlighting the proinflammatory activity of BK, Riedl described BK as “… part of the inflammatory milieu… when activated, B2R triggers oedema, vascular permeability, inflammation, and vasodilation.”
BK signalling is primarily mediated through the activation of the B2R.15 By promoting vascular permeability and inflammation, dysregulation of BK production can contribute significantly to a range of inflammatory disorders, as observed in conditions such as AE-BK,15,17 asthma,16,18 and a cold-induced urticarial syndrome.19 Consequently, B2R represents a potential therapeutic target for disorders that are characterised by BK-mediated hyperpermeability and inflammation.6,20,21
Bradykinin B2 Receptor Antagonism as a Therapeutic Target: Studies and Case Reports
Riedl navigated a series of clinical trials which assessed B2R antagonism in BK-mediated conditions such as HAE. For example, in HAE, a type of AE-BK, B2R antagonism has recognised therapeutic effects on disease manifestations in an acute, on-demand treatment (ODT) setting.6,19,22,23 In patients with chronic asthma, B2R antagonism versus placebo has been shown to improve pulmonary function test performance (e.g., forced expiratory volume in 1 second and peak expiratory flow rate) by 10% after 4 weeks.24 However, Riedl noted, despite improvement in measured pulmonary function, patients reported no significant clinical benefit in symptom score or reduction in rescue medication when compared to placebo.24 B2R antagonism has also been shown to reduce fatigue in FXII-Associated Cold Autoinflammatory Disease (FACAS).19 Riedl concluded that, whilst BK is implicated in a range of inflammatory disorders, such as AE-BK, further research is needed to delineate the specific pathologies in which BK is a core mediator from those where it acts as a conditional contributor alongside other factors.
Patients with acute ischaemic stroke (AIS) receive thrombolytic/fibrinolytic therapy to improve blood flow.25 However, a potential side effect of tissue plasminogen activator (tPA) fibrinolytic treatment in some patients with AIS is orolingual and facial angioedema.26,27 tPA-induced angioedema provides a valuable model to assess the effects of B2R antagonism during an ischaemic condition.27-31 In a series of case reports, patients with AIS and tPA-induced angioedema were administered B2R antagonist treatment. B2R antagonism appeared to reduce angioedema progression, with no reported adverse effects of B2R antagonism on the evolution of AIS.29-33
A randomised, double-blind, placebo-controlled crossover study (N=11) investigated the effects of B2R antagonism in patients experiencing intradialytic hypotension (IDH) during haemodialysis, hypothesised to occur as a result of an increased production of vasodilators, such as BK.34 Of the 11 patients undergoing haemodialysis, seven experienced IDH, defined as a reduction of systolic blood pressure ≥20 mmHg during haemodialysis. A post-hoc stratified analysis based on the presence of IDH revealed that B2R antagonism rescued the drop in blood pressure in patients with IDH but had no effect in patients without IDH. Further, B2R antagonism did not affect heart rate and had no associated adverse events.34
The efficacy of B2R antagonists for the treatment of angiotensin-converting enzyme (ACE) inhibitor-induced angioedema (AE-ACEI) has been assessed across randomised controlled trials. Within 10–12 hours after the onset of AE-ACEI, patients were administered 30 mg of B2R antagonist subcutaneously. Although the primary endpoint of time to symptom resolution or meeting discharge criteria was not met in two of the three studies cited, B2R antagonism did not result in increased blood pressure, and no serious adverse events were reported.35-37
Riedl outlined a clinical case of a patient with HAE observed under repeated B2R antagonism in the form of repeated, acute treatment for a total of 141 HAE attacks over 3 years. No systemic or cardiovascular side effects from this repeated on-demand treatment were reported.38 Considering these studies, Riedl noted that, according to the evidence available to date in humans, the role of B2R in overall cardiovascular homeostasis may be conditional, though data from additional human studies will be important.
Unmasking Bradykinin-Mediated Angioedema: Connecting Evidence, Innovation, and Unmet Needs
AE is a heterogeneous condition with variability across its aetiology, pathophysiology, frequency, and symptomology.39 Cohn explained that “recurrent AE is a field of many unmet needs, and even the classification of the disease itself is challenging.” Reflecting this complexity, he highlighted that several taxonomic systems are currently in use amongst clinicians, each with varying proposed classifications of AE. Cohn emphasised that exact classifications of AE have some inconsistencies across different guidelines and consensus documents, with taxonomical gaps remaining and updated guidelines becoming increasingly necessary as novel genetic mutations are uncovered.39
In 2021, updates to the International World Allergy Organization (WAO)/European Academy of Allergy and Clinical Immunology (EAACI) guidelines incorporated published evidence to support clinicians and patients in making informed disease management decisions for HAE.19 More recently, the Definition, Acronyms, Nomenclature, and Classification of Angioedema (DANCE) framework was developed to provide a global consensus, integrating clinical expression, underlying mechanisms, biomarkers, and genetics to define different subtypes of AE (Figure 2).39 Under the DANCE classifications, AE-BK encompasses hereditary C1 esterase inhibitor (C1INH) deficiency (HAE-C1INH), acquired C1INH deficiency (AAE-C1INH), and hereditary angioedema with normal C1INH and with genetic mutations in genes encoding for elements of the KKS.39 Two newly described forms of HAE, HAE-CPN (HAE-carboxypeptidase N) and HAE-DAB2IP (disabled homolog 2 interacting protein), are considered to be at least in part BK-mediated, but where these fit into the DANCE classification is not yet clear.41

Figure 2: The DANCE classification framework for angioedema.39
*Also designated HAE-nC1INH.
†The role of BK as mediator of AE symptoms seems to be indirect or conditional.
Adapted from Reshef et al.39
AAE-C1INH: acquired angioedema due to C1INH deficiency; ACEI: angiotensin-converting enzyme inhibitor; AE: angioedema; ANA: anaphylaxis; ANGPT1: angiopoietin-1; BK: bradykinin; C1INH: C1 esterase inhibitor; CPN: carboxypeptidase N; DAB2IP: disabled homolog 2 interacting protein; DANCE: Definition, Acronyms, Nomenclature, & Classification of Angioedema; DI: drug-induced; DPPIV: di-peptidyl peptidase IV blocker; EAE: episodic angioedema with eosinophilia; FXII: factor XII; HAE: hereditary angioedema; HAE-C1INH: hereditary angioedema due to C1INH deficiency; HAE-nC1INH: hereditary angioedema due to normal C1INH; HSST: heparan sulfate 3-O-sulfotransferase 6; KKS: kallikrein-kinin system; KNG: kininogen; MC: mast cell; MYOF: myoferlin; NSAID: nonsteroidal anti-inflammatory drug; PLG: plasminogen; SCLS: systemic capillary leak syndrome; tPA: tissue-type plasminogen activator; UNK: unknown; URT: urticaria; VE: vascular endothelium (dysfunction).
Cohn explained that there are several types of AE-BK, which encompass all AEs attributed to the KKS cascade and BK production.39 C1INH is believed to regulate BK production by inhibiting FXIIa, which activates prekallikrein to kallikrein, and by directly inhibiting plasma kallikrein, which cleaves HK into BK.39 Reduced levels or dysfunction of C1INH result in excess BK.40,42 AEs mediated by BK include HAE-C1INH, forms of HAE-C1INH, and acquired angioedema due to C1INH deficiency. HAE-C1INH is inherited, mediated by deficient or dysfunctional C1INH,19,39,40 resulting in excess BK production.40 AAE-C1INH is caused by deficiencies in C1INH, as a result of pathomechanisms that include consumption or anti-C1INH antibodies, typically acquired secondary to autoimmune or haemato-oncologic conditions (mainly lymphoma). However, in selected cases, no underlying condition can be identified.40
AE-BK also includes some forms of HAE with normal C1INH (HAE-nC1INH), which have a similar phenotype to HAE-C1INH, but with normal C1INH inhibitor levels, and can be classified based on the underlying genetic cause.39 Within the DANCE framework, there are four established forms of HAE-nC1INH associated with genetic variants of HAE-nC1INH (FXII, plasminogen, and kininogen), all mediated through the activation of B2R.
Several other AE types have been proposed, for which there is debate around the role of BK: AE due to vascular endothelium dysfunction, and drug-induced AE.39 The latter encompasses all drug-induced AEs, and the first includes AEs believed to be related to intrinsic vascular endothelium abnormalities and variants in genes encoding for elements involved in the regulation of endothelial functions. The role of BK in at least some of these types has not been excluded.39
HAE-unknown type (HAE-UNK) comprises a population of patients with a phenotype indicative of HAE-nC1INH, for which the cause is unknown or not associated with known genetic variants, some of which are believed to be mediated by BK.39,43 An area of ongoing research, diagnosing HAE-UNK can prove challenging and difficult to identify.39,41 Possible causes of HAE-UNK may be BK-mediated, vascular endothelium dysfunction-mediated, or unknown in aetiology and mechanism.39 Diagnosing HAE-UNK involves a positive family history regarding AE and exclusion of known causes of recurrent AE, including C1INH deficiency, mast cell involvement, genetic variants, and associated medication use.43
Differential diagnosis between recurrent AE subtypes and within HAE-UNK remains complex and time-consuming, with efforts ongoing to identify measurable biomarkers that facilitate the diagnosis of AE-BK and AE subtypes, thereby identifying patients likely to be responders to treatment specific to AE-BK.44
Barriers to Diagnosing Bradykinin-Mediated Angioedema
Cohn emphasised how the use of diagnostic biomarkers could shorten the time to diagnosis, as well as enable patient-specific management, through the identification of patients who may respond better to certain treatments.
Moreover, current access to testing procedures varies between clinics, causing delays in diagnosis.19,45-47 Pathway-specific testing is limited, with current diagnostic indicators unable to distinguish between AE-BK and other AE subtypes, or between AE and other inflammatory conditions.46 For assays that target KKS, analytical challenges include analyte instability and susceptibility to pre-analytic ex vivo activation.48
Cohn summarised that “Achieving a diagnosis is a slow process, and there can be huge delays. There are access barriers; we need pathway-specific testing to identify BK-mediated AEs… However, this is challenging: BK has a very short half-life and is very hard to measure. There is a lot of work to be done in this field.”
Current Expert Consensus
Currently, a diagnosis of HAE-nC1INH is based upon clinical criteria facilitated by expert consensus (Figure 3).43,50,51 Despite this, the diagnostic journey remains a challenge due to a lack of standardised diagnostic approaches and objectively measurable biomarkers for different subtypes of AE.43,52

Figure 3: Algorithm for the diagnosis of angioedema.43
*Family history may be an unreliable marker of HAE.
Adapted from Zuraw et al.43
AE: angioedema; ACE: angiotensin converting enzyme; C1INH: C1 esterase inhibitor; HAE: hereditary angioedema; HAE-C1INH: hereditary C1INH deficiency; HAE-UNK: HAE-unknown type; LTP: long-term potentiation; NSAID: non-steroidal anti-inflammatory drugs.
Unmet Needs in Bradykinin-Mediated Angioedema Management
Despite the availability of treatment options, further progress is needed in terms of controlled trial data for HAE-nC1INH, particularly in subtypes without defined genetic variants.43,53 Additionally, data from clinical trials are limited for acquired angioedema due to C1INH deficiency (AAE-C1INH).54
Goals for ODT for AE-BK should prioritise efficacy, safety, tolerability, and overall treatment experience.41,55,56 This includes evaluating the efficacy in specific types of angioedema pathologies,57 ensuring early onset and durable response without requiring multiple administrations,58 and minimising tolerability issues related to administration57 and/or unwanted side effects. Additionally, it is important to retain patients’ ability to perform daily activities,58 and decrease their reliance on healthcare resources.57 “We need treatment options that are efficacious, have low issues with tolerability, work fast and have a sustained effect, and are tailored to the treatment preference options of the patient,” said Cohn.
Regarding clinical trials investigating the ODT of HAE attacks, Cohn highlighted the heterogeneity in current clinical outcome assessments measured in previous studies, which limits the comparability of results across different treatment methods. He highlighted a recent initiative, Project AURORA, a Delphi consensus study which set out to establish a Core Outcome Set to be utilised and aligned across future clinical trials.58 The Core Outcome Set delineates five key standard outcomes that are recommended to be used to evaluate outcomes in clinical trials for on-demand treatment of HAE attacks (Figure 4).58,59

Figure 4: Core Outcome Set for the efficacy of acute treatment of hereditary angioedema.58,59
One of the established key outcomes was the time to end of progression, which involves the patient recognising the first effects of ODT, indicating when symptoms have stopped progressing and they have reached a state of stability or already started improving. Cohn added that “the shorter the time to end of progression, the sooner the patient can adjust and regain their daily activities.” Project AURORA achieved a high level of consensus on the core outcome set.58
In terms of developing LTP, both physicians and patients have several goals for the future. These include treatment efficacy across, and for specific types of AE-BK,56,57 minimising attack frequency,56 ideally to reach attack freedom,60 fast achievement of prophylaxis and reliable protection,57 low tolerability issues,19 convenience around handling, accessibility and storage,56,59 and administering treatment to minimise impact on daily living,56,58 decrease the use of healthcare resources,19,61 and empowering patients with AE-BK to achieve disease control and improve their health-related quality of life.19
The Future of Biomarkers
Looking to the future, symposium chair Farkas expressed her hopes for the possibility of precision medicine in AE, echoing her fellow experts in that “we need early and accurate diagnosis to be able to introduce suitable treatment, to establish whether AE is BK-mediated or not. This is a key question that cannot be answered by symptomatology alone; we need established laboratory markers, kinin biomarker assays.”62 Instead of a one-size-fits-all approach for managing AE, she emphasised the need for innovative approaches to diagnosis that can be carried out at the patient’s bedside, and used to personalise treatments which align with each patient’s individual characteristics based on the identification of biomarkers.
Cohn reiterated the need for quick and efficient biomarkers to establish cases of AE-BK, highlighting that in current clinical practice, clinicians may only rely on response to B2R antagonism as a method of diagnosing AE-BK. He noted that this is a poor diagnostic approach, as it can be unclear whether a response is a result of the antagonist or the natural course of the disease.
Conclusion
In conclusion, recurrent AE represents a heterogeneous disease with a diverse range of pathogenetic mechanisms and mediators, notably BK. The challenges surrounding unmet needs regarding treatment efficacy, tolerability, and burden currently remain for the management of AE-BK, encompassing both LTP and ODT. Moreover, the diagnosis, classification, and treatment underscore the need for further research, including the development of standardised guidelines to identify disease subtypes, specific biomarker diagnostic testing, and the alignment of outcomes assessed in future clinical research trials.






