Interview Summary
Generalised myasthenia gravis (gMG) is a rare, chronic IgG-mediated autoimmune neuromuscular disease characterised by fluctuating muscle weakness. Traditional management has relied on acetylcholinesterase inhibitors, steroids, immunosuppressants, and thymectomy, but these approaches are limited by delayed and suboptimal efficacy, significant side effects, and long-term safety risks. After decades without therapeutic innovation and suboptimal treatment options, the treatment landscape has recently transformed with the approval of novel targeted therapies, including neonatal Fc receptor (FcRn) blockers. The initial years following diagnosis of gMG represent a critical window of high disease activity and risk of life-threatening myasthenic crises. Recognition of this early vulnerability has shifted treatment strategies toward earlier use of advanced therapies in patients with high clinical disease activity shortly after diagnosis.
The following discussion is based on an interview conducted by EMJ with Jan D. Lünemann, Professor of Neurology and Neuroimmunology, University of Münster, Germany. This article follows a previous EMJ interview in which Lünemann shared his experience using FcRn blockers, including efgartigimod alfa, discussed treatment goals and outcome measures, and presented a clinical case illustrating remission and improved quality of life with efgartigimod alfa. In this latest discussion, Lünemann described the importance of achieving early minimal symptom expression (MSE) in gMG, noting that while early achievement of MSE is an independent predictor of long-term prognosis, many individuals continue to experience a substantial disease burden while receiving traditional therapies. Lünemann highlighted evidence supporting early, assertive treatment strategies that drive improved efficacy outcomes while minimising reliance on high-dose corticosteroids. Finally, Lünemann reviewed real-world and long-term clinical study data showing that efgartigimod alfa enables rapid and sustained MSE, improves daily functioning, and reduces corticosteroid use compared with standard of care (SoC).
“Based on my own clinical experience of treating gMG patients at our centre, responses to immunotherapy tend to be more favourable when treatment is initiated early after disease onset compared to a later start. Similar observations have been reported in other autoimmune diseases, in which a recognised ‘window of opportunity’ exists early after disease onset, such that earlier initiation of immunotherapy and achievement of disease control is associated with greater long-term clinical benefit,” said Lünemann.
INTRODUCTION
Lünemann described gMG as a complex chronic autoimmune disease that can be managed but not yet cured.1 Standard treatment for gMG has remained largely unchanged for decades and is based on symptomatic therapy with acetylcholinesterase inhibitors, alongside chronic therapy including corticosteroids and steroid-sparing immunosuppressive agents, such as azathioprine.2-4 Other commonly used steroid‑sparing immunosuppressive agents, such as mycophenolate mofetil, methotrexate, and cyclosporine A, are used in clinical practice but remain off-label for gMG in the EU.2,3 These immunosuppressive agents could be associated with delayed onset of action and limited reversibility in case of infections, and their long-term use carries well-recognised safety risks, including malignancies, hepatotoxicity, and cytopenias.2,3 In addition, thymectomy is an established therapeutic option in patients with acetylcholine receptor (AChR) antibody-positive gMG.2,3 However, Lünemann noted that a substantial proportion of patients do not achieve satisfactory clinical improvement with these commonly used therapies,4 and the onset of meaningful clinical efficacy can take up to 1 year.4
“Long-term therapy with corticosteroids may result in significant side effects, and the onset of clinical efficacy of non-steroidal immunosuppressants may take a couple of months to a year,” Lünemann explained.
SIGNIFICANCE OF MSE IN gMG
Lünemann explained how treatment of gMG has changed fundamentally in recent years due to the approval of innovative new therapies, such as complement inhibitors and FcRn blockers, including efgartigimod alfa, as add-on treatment options.2,3 These new approaches have significantly improved disease management in gMG, shifting the therapeutic goal from avoiding life-threatening myasthenic crises to achieving MSE.5,6 Lünemann explained that MSE is characterised by the absence of functionally-relevant limitations in daily life due to myasthenia, and that “achievement of MSE should ideally be associated with optimal tolerability and safety, such that any side effects that occur require no medical intervention. This is the ultimate goal of myasthenia gravis therapy today.”
“In my experience, MSE can only be achieved in around 10–15% of patients using SoC therapies. In contrast, studies with newly approved therapies have shown that this status can be achieved in about 20–40% of patients, even over relatively short observation periods. Furthermore, long-term studies and real-world studies suggest that this percentage can be even higher when used earlier in the treatment paradigm.”
EARLY ASSERTIVE TREATMENT TO ACHIEVE RAPID DISEASE CONTROL IS AN EMERGING STRATEGY IN gMG
Despite their clinical benefit, the earlier use of innovative therapies for gMG is limited in many countries, with a few exceptions.3,7 Lünemann explained that, in Germany, treatments are indeed formally approved for use in all patients with AChR antibody-positive gMG in addition to symptomatic therapy, although a national expert consensus recommends that they be prioritised for patients with the greatest unmet therapeutic need, including those with highly active disease and those with difficult-to-treat gMG.8,9
Lünemann explained that in clinical practice, patients with difficult-to-treat gMG have generally been able to access these newer therapies, whereas patients with highly active disease early after diagnosis have had more limited access. Epidemiological data indicate that gMG is most active and unstable during the first few years following disease onset, a period that is also associated with the highest risk of life-threatening myasthenic crisis.10,11 On this basis, the German expert consensus advocates for the early use of these newer therapies in patients with high clinical disease activity shortly after diagnosis, particularly in those presenting with myasthenic crisis at onset or experiencing recurrent crises.8 Lünemann noted: “Early intervention in these patients may help achieve effective disease control sooner.” This is important, as early disease control is thought to help limit neuromuscular junction damage and preserve functional reserve, while delayed treatment may allow irreversible damage and sustained weakness.12
‘EFT’ APPROACH
The key study that supports early and effective disease control for gMG is the 2023 Japanese Early Fast-Acting Treatment (EFT) study, a prospective real-world study that included over 1,000 patients across multiple gMG centres in Japan.13 The EFT approach involves initiating rapid-acting therapy, such as plasmapheresis, intravenous immunoglobulin, and/or intravenous high-dose methylprednisolone, early in the disease course in order to improve disease control and reduce steroid-dosing requirements as early as possible. The EFT study demonstrated that this approach can achieve earlier disease remission, defined as minimal impact on daily activities, and faster reduction of corticosteroid dosage to 5 mg daily compared to SoC.13 However, due to high risks and numerous side effects, these treatments are typically used cautiously, and are often reserved for the management of myasthenic crisis.8,13 Against this backdrop, targeted therapies, such as efgartigimod alfa, have emerged as a promising option.8 A research group in China recently investigated the real-world impact of using efgartigimod alfa early compared with SoC in daily clinical practice.6 This multi-centre, real-world study evaluated the proportion of patients with myasthenia gravis achieving MSE and the time required to reach it after receiving multiple cycles of efgartigimod alfa treatment.6
Note: The Chinese real-world study included patients with muscle specific kinase antibody-positive (MuSK-Ab+) gMG. No sub-analysis of the AChR antibody-positive group was performed during the study. In the overall analysis, 4.0% of patients in the efgartigimod alfa group (n=76) had MuSK-Ab+ gMG and 9.7% of patients in the SoC group (n=124) had MuSK-Ab+ gMG.6 Efgartigimod alfa is not approved for use in patients with MuSK-Ab+ gMG.14
NEW REAL-WORLD EVIDENCE ON EARLY EFGARTIGIMOD ALFA USE VERSUS TRADITIONAL THERAPIES
The study noted that early MSE is an independent predictor of long-term prognosis,6 and therapies that are capable of rapidly and effectively controlling disease activity are, therefore, of particular interest. In a recent real-world observational analysis, rates of MSE and time to MSE were assessed in 200 patients with gMG at 16 hospitals in Southern China where efgartigimod alfa was introduced earlier in the treatment course (median disease duration: 1 [0.3–4.5] year) and after patients were already receiving SoC (non-steroidal immunosuppressive therapy, corticosteroids, or combination therapy).6 The study included 76 patients treated with efgartigimod alfa and 124 patients on SoC alone. Efgartigimod alfa was shown to achieve a significantly higher MSE rate compared with SoC (73.3% versus 22.6%; p<0.001) and reach MSE approximately four times faster than SoC alone (0.7 months versus 3.3 months; p<0.001).6 “This study reinforces the value of early biologic intervention, and highlights the progress that has been made with newer therapeutic options, such as efgartigimod alfa, for patients with gMG,” emphasised Lünemann.
Treatment responses were also analysed according to baseline disease severity in this study, comparing patients with higher Myasthenia Gravis Activities of Daily Living (MG-ADL) scores with those with lower scores. In all subgroup analyses, efgartigimod alfa showed a higher probability and shorter time to achieving MSE than SoC alone, regardless of initial disease severity, demonstrating the benefit of treatment across a broad spectrum of baseline disease severities.6
From a mechanistic perspective, Lünemann observed that the magnitude of clinical improvement appears to depend less on baseline MG-ADL score and more on whether the chosen therapy effectively targets the predominant pathogenic mechanism in an individual patient. FcRn blockers, such as efgartigimod alfa, reduce circulating pathogenic IgG by targeting and blocking FcRn.15 Lünemann explained that if the amount of pathogenic IgG-mediated processes are primary drivers of disease activity, therapies that reduce circulating IgG levels may result in marked improvements in daily functioning.
Lünemann also noted that high rates of MSE appear to be achievable across different classes of innovative therapies for gMG, including both FcRn blockers and complement inhibitors,16,17 suggesting that effective disease control can be attained through multiple therapeutic mechanisms. Physicians and patients should jointly evaluate the risk/benefit ratio of the different options when making a treatment selection.
LONGER-TERM DURABILITYOF RESPONSE WITH EFGARTIGIMOD ALFA
ADAPT is currently the only published Phase III, placebo-controlled trial in gMG to demonstrate a significantly higher rate of MSE compared with placebo, with 40% of patients treated with efgartigimod alfa achieving MSE versus 11% in the placebo group (p<0.001).18 ADAPT and its open-label extension (OLE), ADAPT+, together represent 3 years of clinical experience.19 In this context, Lünemann explained that it is also important to assess the longer-term durability of response with advanced treatments for gMG. ADAPT-SC+ was a Phase III OLE study of the pivotal ADAPT-SC study in patients with gMG.20 ADAPT-SC and its OLE, ADAPT-SC+, together represent 459.4 patient-years of follow-up and up to 33 treatment cycles.20 The primary objective was to evaluate the long-term safety and tolerability of subcutaneous efgartigimod alfa, while efficacy outcomes were also assessed.21 The study demonstrated significant and clinically meaningful improvements in mean MG-ADL total scores, with rapid benefits observed as early as Week 1 and sustained through Week 163 (over 3 years;
Figure 1).20 In addition, patients treated with efgartigimod alfa maintained high rates of MSE across multiple treatment cycles, with approximately 60% of participants achieving MSE at any time during the study.21 Importantly, no new safety signals were identified during the OLE phase.20 Lünemann commented that these findings demonstrate the effectiveness of the targeted mechanism of action of efgartigimod alfa in achieving MSE, and, together with recent Chinese real-world data, support the principle that efgartigimod alfa can provide effective and sustained control of disease activity.
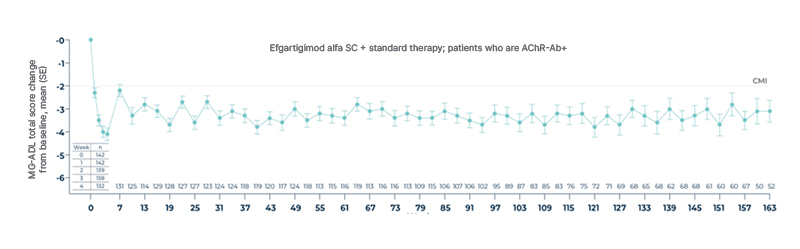
Figure 1: ADAPT-SC+: Mean change from baseline in total MG-ADL score over time.*20
*Mean (SD) study duration for all participants was 932 (333) days. A total of 138 (76.7%) participants completed the study and 42 (23.3%) discontinued early, most commonly due to withdrawal of consent (n=14 [7.8%]), physician decision (n=6 [3.3%]), TEAE (n=5 [2.8%]), and lack of efficacy (n=5 [2.8%]).20
n represents the number of participants with available data per analysis set; follow-up trended toward smaller sample sizes, mainly due to participants completing the study at varying times (since patients rolled over to the study over time) and early discontinuations.20
Adapted from Vu T et al.20
AChR-Ab+: acetylcholine receptor antibody-positive; CMI: clinically meaningful improvement; MG-ADL: myasthenia gravis activities of daily living; SC: subcutaneous; SD: standard deviation; SE: standard error; TEAE: treatment
emergent adverse event.
“Based on my own clinical experience of treating gMG patients at our centre, responses to immunotherapy tend to be more favourable when treatment is initiated early after disease onset compared to a later start. Similar observations have been reported in other autoimmune diseases, in which a recognised ‘window of opportunity’ exists early after disease onset, such that earlier initiation of immunotherapy and achievement of disease control is associated with greater long-term clinical benefit.”
CONCLUSIONS
Although achieving early MSE is an independent predictor of long-term prognosis according to a key real-world study,6 many individuals with gMG continue to experience a substantial disease burden while receiving traditional therapies.22-24 Early assertive treatment strategies that do not rely on high-dose corticosteroids, such as the use of FcRn blockers, are increasingly recognised as critical for achieving symptom control, minimising disease manifestations, and protecting the neuromuscular junction without the significant safety-related trade-offs often made with traditional therapies, such as corticosteroids or non-steroidal immunosuppressants.25
In a real-world analysis of patients treated with efgartigimod alfa within 1 year of disease onset, a significantly greater proportion of patients achieved MSE compared with SoC, and did so approximately four times faster.6 Notably, higher rates of early MSE with efgartigimod alfa were observed regardless of baseline MG-ADL score, including among patients with MG-ADL scores greater than 11.6 These high MSE rates were accompanied by reductions in high-dose corticosteroid use compared with baseline, as well as corticosteroid discontinuation.6
Furthermore, findings from the ADAPT-SC+ OLE study demonstrated that efgartigimod alfa was associated with stable and sustained improvements in daily activities over a 3-year period, together with sustained rates of MSE across multiple treatment cycles.20 Lünemann concluded: “These findings reinforce the value of efgartigimod alfa in delivering rapid, efficient, and sustained control of disease activity.”
| Adverse events can be reported to [email protected] or by using the local medical information telephone number. You will find these in the package leaflet of the SmPC. Adverse event reporting details can also be found here. |




