Meeting Summary
Spinal muscular atrophy (SMA) is a neuromuscular disorder characterized by loss of motor neurons in the spinal cord and brain stem, leading to progressive muscle weakness and atrophy. Three therapies aiming to increase survival motor neuron (SMN) protein are now approved for SMA and have helped to reshape the treatment landscape in terms of survival and disease trajectory. This article discusses data from two recent poster presentations and one manuscript, evaluating the ongoing burden of illness in patients with SMA treated with SMN-targeted treatments. Collectively, these data highlight a residual disease burden that persists in patients with SMA despite treatment, underscoring the need for new therapeutic options to address ongoing muscle weakness and improve motor function.
Background
SMA is a genetic neuromuscular disease resulting in irreversible degeneration of motor neurons and progressive muscle atrophy and weakness due to a deficiency of SMN protein.1,2 It affects approximately one in every 10,000–15,000 live births.3
Three targeted treatments are currently approved in the USA for individuals with SMA from newborn through to adulthood. Nusinersen, risdiplam, and the one-time gene therapy onasemnogene abeparvovec-xioi all act by increasing SMN protein levels to support motor neuron survival.3-5
These SMN-targeted treatments have revolutionized the treatment of SMA, slowing the natural progression of the disease and improving motor function in treated patients.6,7 However, many patients with SMA continue to experience persistent disease burden despite treatment.3
Disease Burden in Patients with Spinal Muscular Atrophy Treated with Survival Motor Neuron-Targeted Treatments: A Targeted Literature Review
A targeted literature review and gap analysis in SMA exploring the current clinical burden in treated patients was presented at the Muscular Dystrophy Association (MDA) Conference 2024 by Diana Castro, the Director of the Neurology and Neuromuscular Care Center in Denton, Texas, USA.8
Previously published systematic literature reviews (SLR) in SMA were analyzed, and a targeted search performed on key topics found it to be underrepresented in the current literature. Search parameters were population (SMA), treatment type (all), publication date (2019–2023), geographic region (all), and language (English). An assessment of unmet needs related to motor function and muscle strength was then conducted in order to characterize the disease burden in patients with SMA treated with SMN-targeted treatments. From 14 identified SLRs, a total of 42 out of 500 publications were included in the final assessment.8
This up-to-date review of the current literature identified significant unmet needs associated with an ongoing clinical disease burden in patients with SMA.8 Despite treatment, clinical improvement varied for adults and pediatric patients with either Type 2 or 3 SMA treated with nusinersen over a 10- to 14-month period, with change in Hammersmith Functional Motor Scale–Expanded (HFMSE) score ranging from −1 to +6 points.5 Importantly, parents and caregivers expressed the desire not only for meaningful improvements, but also for the continued development of nuance-sensitive scales used to assess motor function in SMA.8
This research also highlighted persistent unmet needs in SMA, particularly the short- and long-term improvement and sustainment of muscle strength and motor function, as well as the management of fatigability. However, current data on increased motor strength and its impact on motor function remain limited, with only one publication addressing this important issue.8
Important unmet needs associated with patient activities of daily living (ADL) and quality of life (QoL) were also revealed in this literature review. Qualitative and quantitative research showed that SMA is associated with burdens in mobility and walking, as well as respiratory and bulbar features of the disease, such as breathing difficulties, choking, and swallowing. SMA was also shown to impact patient ADLs in areas such as personal hygiene, grooming, dressing, toileting, feeding, transferring, and ambulating. In terms of the existing scales used to assess motor function, patients believed that, although HFMSE and Revised Upper Limb Module (RULM) items were important, they were not sufficiently sensitive to capture the small changes that are seen after initiation of SMN-targeted treatments.9 Echoing this point, clinicians, patients, and caregivers all emphasized the importance of small improvements in SMA, including those that may be perceived by the general population as too small to be beneficial.9 Patients noted that trials should assess changes in abilities related to independence through everyday examples, such as opening doors, combing hair, turning book pages, and holding a drink, for example.9
Overall, this literature review highlighted the need for improvements in three key areas within the current SMA treatment landscape: daily activities (seven publications), fatigue (four publications), and QoL (14 publications; Figure 1).

Figure 1: Identified spinal muscular atrophy literature gaps.8
In conclusion, this targeted literature review demonstrates that, despite advancements in SMA management, significant unmet medical needs remain for treated patients, including the need to address muscle weakness and fatigue, and improve patient ADLs and QoL. The current body of literature likely underestimates the true impact of the disease on patient functional status, fatigue, ADLs, and health-related QoL. Moving forward, additional research is warranted to further describe and document these important measures of residual disease burden in patients with SMA treated with SMN-targeted treatments.8
Characterization of the Longer-Term Effectiveness of Survival Motor Neuron-Targeted Treatments for Spinal Muscular Atrophy: A Systematic Literature Review
Despite the efficacy of SMN-targeted treatments, many individuals with SMA continue to experience persistent muscle weakness and an ongoing SMA-related burden of illness.6,7,10,11 Further data evaluating the long-term impact of these treatments are therefore needed.
Against this backdrop, Jena Krueger, Pediatric Neurologist, Helen DeVos Children’s Hospital, Grand Rapids, Michigan, USA, and colleagues undertook an SLR that aimed to plot the trajectory of longer-term outcomes for patients receiving SMN-targeted treatments (nusinersen, onasemnogene abeparvovec-xioi, and risdiplam). Results for nusinersen, which had the largest body of literature evidence available, were presented at Cure SMA 2025.12
This SLR focused on studies reporting motor function endpoints conducted in the USA, Europe, and Australia between 2017–2024, with special attention to those studies with outcomes beyond 16 months. The primary outcome measures were HFMSE and RULM scores. In total, 120 articles (including 18 clinical trials and 102 real-world studies) primarily evaluating Types 2 and 3 SMA met the criteria for inclusion in the SLR. Of these, 44 publications reported HFMSE scores and 36 reported RULM scores, with 16 and 12 publications, respectively, reporting a median change from baseline at two or more timepoints with at least one assessment beyond 12 months.13-30
The gain in HFMSE scores varied between the different nusinersen studies evaluated in this SLR. The greatest change from baseline in HFMSE score for any single study was 10.8 points, occurring 38 months after treatment initiation. Although the maximum score possible on the HFMSE scale is 66 points, the average maximum total HFMSE score across all nusinersen studies was only 31.7 points, suggesting significant levels of disability among patients.12 A lower HFMSE score indicates greater motor impairment.
Comparison of the HFMSE scores from the CHERISH/SHINE extension study (the only published study with longitudinal data beyond 42 months at the completion of the SLR) with the best-fit curves of the other clinical trials and real-world studies illustrated a consistent trajectory of change following nusinersen initiation. The improvement in HFMSE scores with nusinersen occurred primarily within the first 2 years of treatment, after which the rate of improvement attenuated. Notably, the results indicated a progressive decline in HFMSE scores after 42 months of nusinersen treatment that continued to 92 months (Figure 2). However, this rate of decline was still lower than would be expected in untreated individuals.12

Figure 2: Individual best-fit curves plotted against longer-term data (CHERISH/SHINE) and natural history data.12
The individual curves are best curve fits using polynomial (second- and third-order, centered and non-centered), exponential, or log normal. Longer-term data from Finkel et al.28 were plotted using a third-order polynomial (cubic) with 95% confidence band, and natural history data from Coratti et al.17 were plotted using a second-order polynomial (quadratic). A lower HFMSE score indicates greater motor impairment.
Adapted from Krueger J et al.12
HFMSE: Hammersmith Functional Motor Scale–Expanded.
Gains in RULM scores varied between nusinersen studies, with the greatest change from baseline for any single study being 9.3 points at 30 months post-treatment initiation. Across all studies, the average maximum total RULM score in treated patients was 22.7 points out of a possible total of 37. RULM scores demonstrated a rapid initial improvement in motor function that occurred in the first 2–3 years following nusinersen initiation, followed by a plateau with limited or no further gain.12 A lower RULM score indicates greater disability in terms of upper limb performance.
Overall, these longer-term clinical trial data show that, after initial improvements, there is a plateau in gains for motor function, as assessed by HFMSE and RULM scores, approximately 2 years following nusinersen initiation. Although outcomes are significantly improved compared with natural history, notable residual disability and unmet clinical needs persist in treated patients, as evidenced by the degree of motor function attained and the progressive decline in HFMSE scores after 42 months of nusinersen therapy. Collectively, these data highlight the need for additional therapeutic approaches that can further improve motor function and prevent long-term decline in treated patients.12
Remaining Burden of Spinal Muscular Atrophy Among Treated Patients: A Survey of Patients and Caregivers
A recent cross-sectional USA survey, led by Julie Parsons, Codirector of the Neuromuscular Clinic, Children’s Hospital Colorado, Aurora, USA, set out to explore in greater depth how this residual disease burden in SMA impacts patients and their caregivers.3
This web-based survey was conducted in August 2024 among non-ambulant patients with SMA currently receiving risdiplam or nusinersen and their primary carers. Survey questions captured the clinical, humanistic, productivity, and caregiver-related burden of disease, while the Patient-Reported Outcomes Measurement Information System (PROMIS) Fatigue and EuroQoL 5-Dimension 5-Level Scale (EQ-5D-5L) tools were used to assess fatigue and QoL.3
In total, 40 pediatric patients represented by caregiver proxies and 68 adult patients took part in the survey. The majority of patients (82.5% of pediatric and 94.1% of adult patients) had been on an SMN-targeted treatment for 2 years or more and nearly half (45.0% and 48.5%, respectively) had been on treatment for 4 years or longer. Despite continued treatment, muscle weakness was reported in 95% of pediatric and 100% of adult patients. The majority, 63% and 68% respectively, described this muscle weakness as “severe” or “very severe.” Notably, muscle weakness was strongly attributed to patients’ difficulties in performing daily activities and to limitations in motor function. In addition to its substantial impact on motor function and performance of ADLs, muscle weakness was also linked to worse overall health among treated patients with SMA. Adult patients with severe or very severe muscle weakness reported significantly lower overall health scores, as assessed by the EuroQol Visual Analogue Scale (EQ-VAS), than those with moderate or mild muscle weakness. This broad impact of muscle weakness in patients with SMA highlights the importance of addressing this key symptom as part of an overarching disease management strategy.3
Despite the significant muscle weakness revealed in this survey, overall fatigue levels were lower than expected, potentially due to patients intentionally limiting their everyday activity. Mean PROMIS scores fell within the moderate range among pediatric patients and within the mild range for adult patients. Nevertheless, increased fatigue was still associated with worse overall health among treated patients.3
Consistent with the importance of small but meaningful motor improvements highlighted in the targeted literature review, both adult and pediatric patients in this survey placed most value on being able to independently complete activities requiring fine motor skills. The top three activities that patients prioritized most were using everyday tools with push buttons (e.g., remote controls), using their hands (e.g., handling utensils), and using their fingers (e.g., collecting small food items).3
SMA was also found to impact patient QoL, well-being, and mental health as evaluated by the EQ-5D-5L and its respective proxy version. Most adult patients (72%) reported some level of anxiety or depression, while pain or discomfort was experienced by 68% and 84% of pediatric and adult patients, respectively. The three most burdensome impacts of SMA, as rated by patients of all ages, were difficulties performing daily activities independently, followed by limited physical functioning, and restricted ability to participate in social events.3
Several key unmet needs were also identified among surveyed patients undergoing treatment with SMN-targeted treatments. Both pediatric and adult patients cited mobility limitations (75% and 69%, respectively) and muscle weakness (53% and 56%, respectively) as being the aspects of SMA least improved by their current treatment (Figure 3). These ongoing issues of muscle weakness and mobility limitations were reported to have “very much” of an impact on QoL by 57% of pediatric patients and 42% of adults living with SMA.3
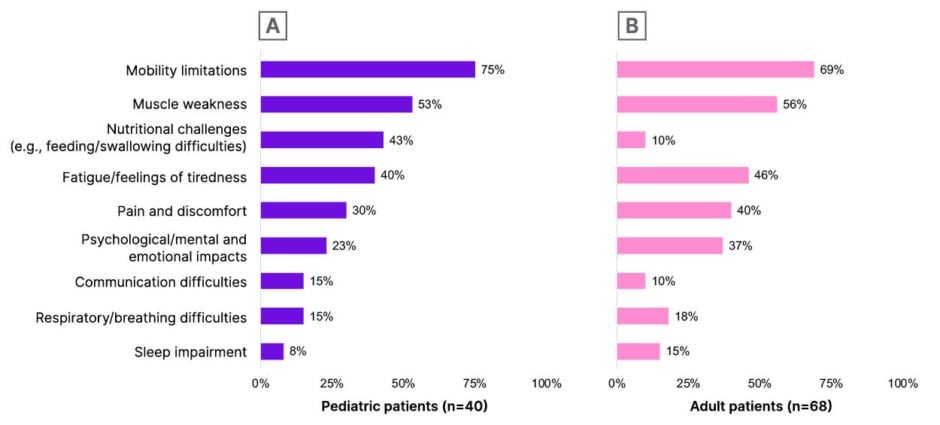
Figure 3: Spinal muscular atrophy symptoms least improved with current survival motor neuron-targeted treatment.3
A) Pediatric patients (n=40). Pediatric patient data was reported by caregiver proxies. B) Adult patients (n=68). Adult patient data was self-reported. Participants were asked to select the three impacts the patient considered/caregivers thought the patient with SMA considered to be least improved or helped by the patient’s current treatment.
Adapted from Parsons J et al.3
SMA: spinal muscular atrophy.
The survey also uncovered an impact on productivity in terms of missed school attendance and work absenteeism among patients with SMA, while caregivers also reported a burden that affected their QoL and employment. Most caregivers of pediatric and adult patients felt some level of anxiety/depression (70% and 90%, respectively), and around half said they had had to reduce their work hours to accommodate caring for a person with SMA.3
In summary, the results of this survey highlight that, despite current SMN-targeted treatments, patients with SMA continue to experience muscle weakness that impacts motor function and the ability to perform daily activities, which reduces their overall health and QoL. Notably, muscle weakness and mobility limitations were reported as the SMA symptoms least improved by currently used SMN-targeted treatments. The most burdensome symptoms described by patients, along with the activities they most valued being able to perform independently, were also impacted by this ongoing muscle weakness. Overall, these results underpin the need for new management approaches for SMA that focus on increasing muscle strength and promoting motor function, thereby leading to improvements in patient ADLs, QoL, and other key humanistic aspects of SMA care.3
Conclusion
Collectively, these recently presented and published data show that, despite the efficacy of SMN-targeted treatments, patients with SMA continue to experience a residual disease burden and substantial unmet treatment needs remain.3,8,12 Additional therapeutic approaches are therefore needed that can enhance muscle strength and improve motor function in patients with SMA, thereby preventing long-term decline and optimizing patient care in the future.3,12





