Summary
Despite advances in first-line endocrine therapy (ET) plus cyclin-dependent 4/6 kinase inhibitors (CDK4/6i) in oestrogen receptor-positive, human epidermal growth factor receptor 2-negative (ER+/HER2-) metastatic breast cancer (mBC), disease progression often occurs. When this happens, most patients remain eligible for ET with or without targeted therapies, while those with visceral crisis or primary endocrine resistance (progression after <6 months of ET plus CDK4/6i) should be treated with cytotoxic regimens (chemotherapy or antibody–drug conjugates). This review discusses the latest advancements in overcoming resistance to first-line ET in ER+/HER2− metastatic breast cancer that emerged during 2024 from the American Society of Clinical Oncology (ASCO) Annual Meeting in May and June, the European Society for Medical Oncology (ESMO) Congress in September, and the San Antonio Breast Cancer Symposium in December. The article highlights the crucial role of biomarker-driven strategies to address the emergence of ESR1 mutations and the presence of intrinsic PI3K/AKT/mTOR pathway alterations. Elacestrant is the only oral selective oestrogen receptor degrader (SERD) approved for ESR1-mutated mBC, having demonstrated a hazard ratio (HR) of 0.55 (95% CI: 0.39–0.77) versus standard of care (SOC) in patients with ESR1-mutated tumours in the EMERALD study. An exploratory subgroup analysis of patients with longer prior ET plus CDK4/6i exposure (at least 12 months) demonstrated a median progression-free survival (PFS) of 8.6 months versus 1.9 months with SOC (HR: 0.41; 95% CI: 0.26–0.63), and 5.5 months in patients with co-existing PIK3CA mutations. These findings were supported by real-world studies, which showed a median PFS of 8–9 months with second- and third-line elacestrant, and 5.2 months in patients with co-existing ESR1 and PIK3CA mutations. Additionally, CAPItello-291 demonstrated a 5.5-month median PFS with capivasertib plus fulvestrant in PIK3CA/AKT1/PTEN-altered mBC in patients previously treated with CDK4/6i. Biomarker-driven treatment selection is now critical, emphasising the need for ESR1 mutation testing after each progression.
Primary Resistance to First-Line Metastatic Breast Cancer Treatment Poses an Unmet Medical Need
Approximately 70% of new breast cancer cases are ER+/HER2-.1,2 Despite the effectiveness of ET and continued expression of ERα, many patients have a lifelong risk of metastatic recurrence.3,4
The standard first-line (1L) treatment for ER+/HER2- mBC is a CDK4/6i (i.e., palbociclib, ribociclib, or abemaciclib) combined with ET.5,6 However, the disease generally progresses after 1L treatment,2 and overcoming resistance to 1L CDK4/6i plus ET is an unmet medical need. In patients in whom disease progression occurred within 6 months of 1L treatment, the DESTINY-Breast06 trial demonstrated a significant PFS benefit with trastuzumab deruxtecan (T-DXd) versus chemotherapy in those with HER2-low mBC who were no longer candidates for continued endocrine-based strategies.7 These findings established T-DXd as a second-line (2L) option for patients with visceral crisis or those with primary endocrine resistance (Figure 1).6-14
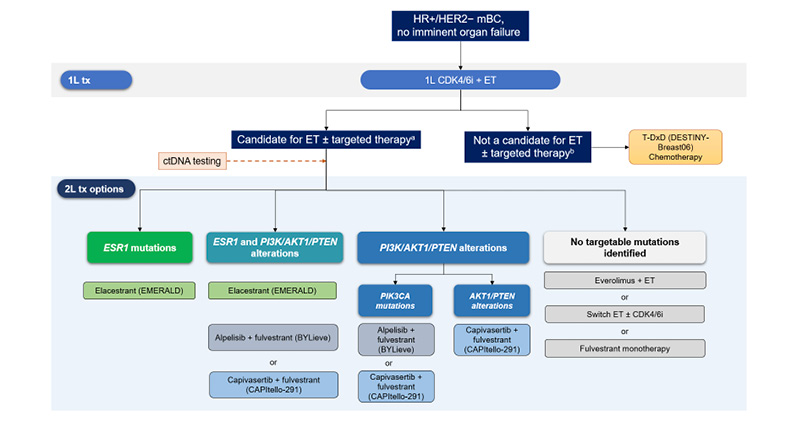
Figure 1: Second-line treatment options for patients with HR+/HER2- metastatic breast cancer.6-14
aExposure to >6 months 1L CDK4/6i, no visceral crisis.
bVisceral crisis, exposure to <6 months 1L CDK4/6i, HER2-low or ultralow mBC.
1L: first-line; 2L: second-line; CDK4/6i: cyclin-dependent 4/6 kinase inhibitor; ctDNA: circulating tumour DNA;
ER+/HER2-: oestrogen receptor-positive, human epidermal growth factor receptor 2-negative; ET: endocrine
therapy; mBC: metastatic breast cancer; T-DXd: trastuzumab deruxtecan; tx: treatment.
Genomic Alterations Determine 2L Treatment Options for Endocrine Therapy-Eligible Patients
Second-line treatment options for patients with ER+/HER2- mBC who remain eligible for ET are largely based on the presence or absence of actionable mutations in the tumour.15,16 Molecular resistance to ET in ER+/HER2- mBC can be intrinsic (present at the onset of oncogenic disease, e.g. BRCA, PIK3CA mutations), or acquired (when mutations develop during or after ET [e.g. ESR1]).2,17
ESR1 mutations are a key driver of acquired resistance to ET, occurring in up to 50% of patients with disease progression after 1L CD4/6i plus ET.8
Additional molecular drivers of mBC include alterations of the PI3K/AKT/mTOR signalling network.18 PI3K/AKT/PTEN signalling can be upregulated either via activating mutations in PIK3CA and AKT1, or by inactivating alterations in PTEN.19 PIK3CA mutations are found in 30–40% of ER+/HER2- mBC, whereas AKT1 and PTEN alterations are found at much lower incidences (around 5–10%).2 ET resistance can also be driven by other alterations in the RAS-MAPK or FGFR1 pathways, as well as by RB1 loss, TP53 inactivation, or increased cyclin E.17
Targeting ER+ Metastatic Breast Cancer with Novel Approaches
Monotherapy
The Phase III EMERALD trial established the efficacy of elacestrant, an oral SERD, compared to SOC fulvestrant or aromatase inhibitor (AI) monotherapy in patients with ER+/HER2- mBC whose disease progressed following prior CDK4/6 inhibitor and endocrine therapy.8,20 Notably, 48% of these patients harboured an ESR1 mutation.14 Elacestrant demonstrated a significant improvement in median PFS with a HR of 0.55 (95% CI: 0.39–0.77) in the ESR1-mutated subgroup and an HR of 0.70 (95% CI: 0.55–0.88) in the overall study population.8
Based on these findings, elacestrant became the first oral SERD to receive regulatory approvals from the U.S. FDA,21 EMA,22 and MHRA23 as a 2L treatment specifically for patients with ER+/HER2-, ESR1-mutated, locally advanced or mBC.
The EMERALD trial included patients with ESR1-mutated mBC with prior exposure to CDK4/6i (100%), chemotherapy (23%), and fulvestrant (24%), and allowed those with primary endocrine resistance to be enrolled.20
Due to the inclusion of such an endocrine-resistant population, an exploratory analysis of the EMERALD data was performed, which suggested a PFS benefit in more endocrine-sensitive patients (prior ET plus CDK4/6i ≥12 months), who had a median PFS of 8.6 months with elacestrant versus 1.9 months with SOC (HR: 0.41; 95% CI: 0.26–0.63).8 The clinically meaningful PFS improvements with elacestrant versus SOC in patients with ESR1 mutations were seen regardless of coexisting mutations (PIK3CA: 5.5 months with elacestrant versus 1.9 months with SOC [HR: 0.42; 95% CI: 0.18–0.94]; TP53: 8.6 with elacestrant versus 1.9 months with SOC [HR: 0.30; 95% CI: 0.13–0.64]).8 The most common Grade 3/4 adverse events (AE) reported in EMERALD were nausea (in 3% of patients), back pain (3%), and increased alanine aminotransferase (2%). Only 6% of patients discontinued elacestrant due to AEs.20
Real-world elacestrant studies are consistent with these findings.9,10 In one independent study (n=756), the time to next treatment was 8.8 months in the 2L setting and 5.9 months in the 3L.9 In patients with co-existing ESR1 and PIK3CA mutations and prior exposure to CDK4/6i plus ET, the median real-world time to next treatment after elacestrant was 5.2 months.9 In a second real-world study (n=276), the real-world median PFS was 8.0 months with 2/3L elacestrant.10 Patients without prior fulvestrant exposure and those receiving earlier-line elacestrant had a longer PFS. These real-world data are consistent with 2/3L elacestrant data in EMERALD, including in those patients with co-existing ESR1 and PIK3CA mutations, underscoring the need for early ESR1 testing and personalised treatment sequencing.
The Phase III EMBER-3 trial compared the oral SERD imlunestrant with SOC ET as monotherapy or in combination with abemaciclib in pretreated patients with ER+/HER2- mBC.24 Unlike EMERALD, EMBER-3 did not require prior CDK4/6i exposure, and it excluded patients previously treated with chemotherapy and fulvestrant. Additionally, the combination arm was added after enrolment into the monotherapy and SOC arms had started. Therefore, between-arm imbalances in baseline characteristics were seen in ESR1 mutation rates and prior CD46i exposure rates.
Imlunestrant monotherapy significantly prolonged PFS versus SOC in patients with ESR1 mutations (5.5 months versus 3.8 months; HR: 0.62; 95% CI: 0.46–0.82; p<0.001); however, a HR of 0.72 (95% CI: 0.52–1.01) was seen in patients with prior exposure to CDK4/6i. The PFS difference was not statistically significant in the overall population (5.6 months versus 5.5 months; HR: 0.87; 95% CI: 0.72–1.04; P=0.12).24 Grade 3/4 AEs occurred in 17% of patients treated with imlunestrant, with the most common being anaemia and neutropenia (each in 2%) and increased aspartate aminotransferase (1%); 4% discontinued due to AEs.24
Vepdegestrant is a novel oral proteolysis targeting chimera (PROTAC) ER degrader. Unlike SERDs, which indirectly cause ER degradation secondary to conformational changes and/or immobilisation, vepdegestrant directly induces ubiquitination of ER, followed by proteasomal degradation.25 Preliminary topline results from the ongoing Phase III VERITAC-2 trial of vepdegestrant versus fulvestrant25 showed that the primary PFS endpoint was met in patients with ESR1 mutations, but not the overall patient population.26
Another investigational therapy, palazestrant, a complete ER antagonist that binds ER and blocks ER-driven transcriptional activity irrespective of ESR1 mutation status, is being evaluated in the Phase III OPERA-01 study.27
Combination Approaches with Selective Oestrogen Receptor Degrader
To overcome additional resistance mechanisms and improve efficacy, elacestrant and imlunestrant were also evaluated in combination with other agents.
Elacestrant combined with abemaciclib was evaluated in patients with ER+/HER2- mBC previously treated with ET and CDK4/6i in ELEVATE and ELECTRA, which included patients with brain metastases.28 Phase I analysis showed the median PFS with this combination was 8.6 months in all patients, 8.7 months in patients with ESR1 mutations, and 7.2 months in patients without ESR1 mutations.28 The safety profile was consistent with the known safety profiles of each agent.
In the imlunestrant plus abemaciclib arm in EMBER-3, the median PFS was 9.4 months, compared with 5.5 months for imlunestrant monotherapy (HR: 0.57; 95% CI: 0.44–0.73).24 The lack of a control arm for imlunestrant plus abemaciclib and the fact that imlunestrant monotherapy did not show significantly improved PFS versus SOC in the intent-to-treat population makes it challenging to contextualise these findings. The incidence of Grade ≥3 AEs was higher with the combination (49%) than with monotherapy (17%), with neutropenia (in 20% of patients), diarrhoea (in 8%), and anaemia (in 8%) being the most common Grade ≥3 toxicities; 6% discontinued the combination due to AEs.24
Recently published data addressed the activity of abemaciclib combined with fulvestrant post-CDK4/6i.29 In the postMONARCH study, the median PFS was 6.0 versus 5.3 months with fulvestrant, with a HR of 0.73 (95% CI: 0.57–0.95) favouring abemaciclib plus fulvestrant.29 In patients previously treated with ribociclib, the HR was 1.01 (95% CI: 0.67–1.51), and in those previously treated with abemaciclib, the HR was 0.66 (95% CI: 0.27–1.64).29
Targeting cyclin-dependent kinase 7 (CDK7) presents another strategy to overcome CDK4/6i resistance. The CDK7 inhibitor samuraciclib combined with elacestrant has an acceptable safety profile and has shown preliminary antitumour activity in the ongoing Phase Ib/II SUMIT-ELA study.30 In the ongoing MORPHEUS BC study, samuraciclib is being investigated in combination with giredestrant, another oral SERD, in patients with disease progression after one or two lines of ET+CDK4/6i.31
Targeting PIK3CA/AKT1/PTEN Tumour Alterations
In the Phase II BYLieve trial, the PI3K inhibitor alpelisib combined with fulvestrant demonstrated a median PFS of 8 months post CDK4/6i plus AI in patients with PIK3CA mutations.11 In the patients with co-existing ESR1 and PIK3CA mutations, the median PFS was 5.6 months.12 The most common Grade ≥3 treatment-related AEs were hyperglycaemia (29%), rash (10%), and diarrhoea (6%); 21% discontinued treatment due to AEs.11 This combination is approved in patients with PIK3CA-mutated ER+/HER2- mBC after progression on ET-based therapy.13
The AKT inhibitor capivasertib combined with fulvestrant was approved in late 202314 as a 2L treatment option for ER+/HER2- mBC after the Phase III CAPItello-291 trial demonstrated prolonged PFS versus fulvestrant in patients with PIK3CA, AKT1, or PTEN alterations.32 The median PFS in the PIK3CA/AKT1/PTEN-altered population who had prior CDK4/6i exposure was 5.5 months (HR: 0.59; 95% CI: 0.48–0.72); the PFS in patients with co-existing ESR1 mutations was not reported.32 The most common Grade ≥3 AEs were rash (12%), diarrhoea (9%), and hyperglycaemia (2%),33 which occurred early (within a median of 8 days) and were manageable with supportive medications, dose adjustment or discontinuation;33 13% of patients discontinued capivasertib due to AEs.33
Strategies for inhibiting multiple signalling nodes of the PI3K pathway include studies with the dual PI3K-mTOR inhibitor, gedatolisib, which was evaluated in combination with palbociclib and fulvestrant in a Phase Ib study in patients with ER+/HER2- mBC after failure of CDK4/6i plus ET.34 The objective response rate was 56% in patients with wild-type or PIK3CA-mutated tumours;34 the Phase III VIKTORIA-1 study of gedatolisib is ongoing.35
Finally, newer generation mutant-selective PI3K inhibitors are in early development, with the hopes of providing more durable responses without the typical AE profiles of non-selective PI3K pathway inhibitors. RLY-2608 plus fulvestrant showed a median PFS of 9.2 months in patients with PIK3CA-mutant HR+/HER2- mBC without concurrent PTEN/AKT alterations.36 AEs were consistent with mutant selective PI3Ka inhibition, with Grade 3 hyperglycaemia reported in 2% and fatigue in 7% of patients, and no Grade 4 or 5 treatment-related AEs.
Biomarker-Driven Optimisation of 2L Therapy
Current guidelines recommend testing for ESR1 mutations in liquid biopsy at each metastatic progression2,37-39 due to their frequent emergence (they have been observed in up to 50% of patients8) following extended ET exposure in mBC (Figure 1). As ESR1 mutations are often subclonal, highly sensitive blood-based circulating tumour (ct)DNA assays are preferred.40 Testing for PI3K/AKT/PTEN pathway alterations is also essential after progression following CDK4/6i therapy to guide the use of targeted agents like PI3K and AKT inhibitors.2,19,40,41
In all-comer populations, the postMONARCH trial showed that continuing CDK4/6 inhibition beyond progression conferred a modest median PFS benefit of 0.7 months versus fulvestrant alone, with a more pronounced benefit in patients previously treated with palbociclib.29
Among patients with PIK3CA, AKT1, or PTEN alterations, capivasertib plus fulvestrant has demonstrated clinical benefit and is now approved for this biomarker-defined subgroup.14,32 However, CAPItello-291 did not report outcomes in patients with co-existing PI3K/AKT/PTEN and ESR1 mutations. Alpelisib also improved PFS in patients with PIK3CA mutations after progression on CDK4/6i plus AI;11 although, resistance was observed in tumours harbouring PTEN or ESR1 mutations.42 Data in patients with co-existing mutations are available only for alpelisib, showing 5.6 months median PFS in the BYLieve trial.12
For patients with ESR1 mutations, both the EMERALD trial8 and real-world studies9,10 support the use of elacestrant monotherapy in the second/third-line setting, particularly in those with prolonged prior CDK4/6i exposure, in whom a median PFS of 8–9 months was observed, and in those harbouring coexisting PI3K alterations, in whom the median PFS was 5.5 months with a manageable safety profile.
| Adverse events should be reported. Reporting forms and information can be found at www.yellowcard.mhra.gov.uk or search for MHRA Yellow Card in the Google Play or Apple App Store.
Adverse events should also be reported to Menarini Stemline UK Ltd via [email protected]. For those in the EU, healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V in the document found here. For those outside the UK and the EU, please refer to the procedures outlined by your local authorities. |