Abstract
Birt–Hogg–Dubé syndrome (BHDS), also known as Hornstein–Knickenburg syndrome, is a rare autosomal dominant disorder caused by mutations in the FLCN gene, characterised by cutaneous lesions, pulmonary cysts, spontaneous pneumothoraces, and renal tumours. The authors report a case of a 23-year-old, previously healthy female who presented with acute left-sided chest pain and was diagnosed with spontaneous haemopneumothorax. Imaging revealed a hydropneumothorax with complete left lung collapse and a breast mass, later confirmed as a fibroadenoma. She also exhibited skeletal features and mitral valve prolapse, raising suspicion of Marfan syndrome, which was excluded through genetic testing. Instead, a pathogenic FLCN mutation (c.1285del, p.His429ThrfsTer39) confirmed BHDS. No renal abnormalities were identified on MRI. This case highlights the diagnostic challenges of BHDS, particularly in the absence of family history or overt cutaneous signs. Prompt recognition and genetic confirmation are crucial for appropriate management and surveillance of associated complications, including renal malignancies and recurrent pneumothoraces.
Key Points
1. Birt–Hogg–Dubé syndrome (BHDS) is a rare autosomal dominant condition caused by FLCN gene mutations, with a triad of skin lesions, pulmonary cysts, and renal tumours.2. This case describes a young female presenting with spontaneous haemopneumothorax and no prior respiratory history, which led to a diagnosis of BHDS. Such presentations are uncommon and highlight the need to consider BHDS in differential diagnoses of spontaneous pneumothorax, especially when associated with other systemic features.
3. Genetic confirmation of an FLCN mutation was pivotal in diagnosing BHDS. This shows the value of molecular diagnostics and the necessity for multidisciplinary surveillance, including renal imaging and pulmonary follow-up, to manage potential complications and screen at-risk individuals effectively.
INTRODUCTION
Birt–Hogg–Dubé syndrome (BHDS) is a rare autosomal dominant genetic disorder caused by mutations in the FLCN gene, which encodes folliculin, a protein involved in regulating several cellular processes, including mTOR signalling and energy homeostasis. First recognised for its distinctive triad of cutaneous lesions, renal tumours, and pulmonary cysts, BHDS has since been shown to present with wide clinical variability, posing significant diagnostic challenges, particularly in patients without a suggestive family history or overt symptoms.1-3
The genetic basis of BHDS was first clarified in the early 2000s. In 2001, Khoo et al.4 mapped the disease locus to chromosome 17p12–q11.2 through a genome-wide linkage study in a Swedish family, while Schmidt et al.5 independently identified a similar region in unrelated families. In 2002, Nickerson et al.6 narrowed this to a 700 kb segment on 17p11.2 and identified FLCN as the causative gene. Schmidt et al.7 later found germline FLCN mutations in 84% of affected families, mostly truncating variants, supporting its role as a tumour suppressor gene. Toro et al.8 confirmed these findings in 88% of cases across 50 families. Expanding to a Japanese cohort, Kunogi et al.9 found FLCN mutations in 69.4% of patients with pulmonary cysts, noting fewer skin and renal features, suggesting ethnic and geographic variability in expression. To date, over 140 unique FLCN mutations have been identified in BHDS.10,11
Here, the authors present a case report on an uncommon initial manifestation of BHDS, spontaneous haemopneumothorax, in a young woman without a suggestive family history or skin findings. It underscores the diagnostic complexity of BHDS in atypical presentations and highlights the critical role of genetic testing in identifying rare hereditary disorders.
CASE DESCRIPTION
A 23-year-old woman with no known comorbidities presented to the pulmonology outpatient department with complaints of left-sided chest pain for 2 days, which had progressively worsened over time. The patient had no complaints of fever, cough, expectoration, or shortness of breath. She denied any history of trauma, smoking, illicit drug use, or other addictions, and had no prior respiratory illness. Her past medical history was notable for prognathism, which had been surgically corrected.
On physical examination, she appeared tall and thin, with long, tapering extremities and a high-arched palate. Oxygen saturation on room air was 96%, and other vital parameters were stable. There were no signs of respiratory distress; however, auscultation revealed decreased breath sounds over the left hemithorax. Cardiovascular examination revealed a mid-systolic murmur, best heard over the left parasternal area. A chest radiograph demonstrated a hyperlucent left hemithorax with an air–fluid level, consistent with a left-sided hydropneumothorax (Figure 1).
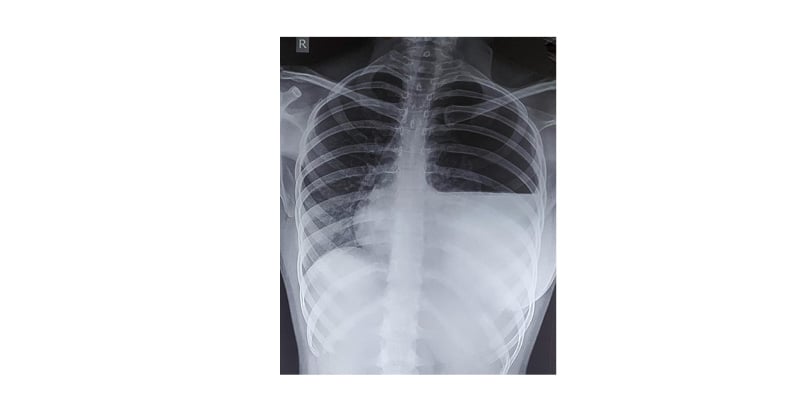
Figure 1: Chest X-ray showing left-sided hydropneumothorax.
The patient was admitted for further evaluation and was managed with surgical intervention in the form of intercostal drain (ICD) insertion for left-sided hydropneumothorax; approximately 100 mL of haemorrhagic fluid was drained. Pleural fluid investigations showed low adenosine deaminase and high lactate dehydrogenase, with numerous/full-field red blood cells (Table 1). Pleural fluid haematocrit was 14.5%. Initial laboratory investigations revealed anaemia (haemoglobin: 7.3 g/dL; haematocrit: 25%). The white blood cell count was elevated at 13,460/μL, suggestive of a possible inflammatory or infectious process. Platelet count was within normal limits (252,000/μL). Renal function test, liver function tests, and serum electrolytes were normal.
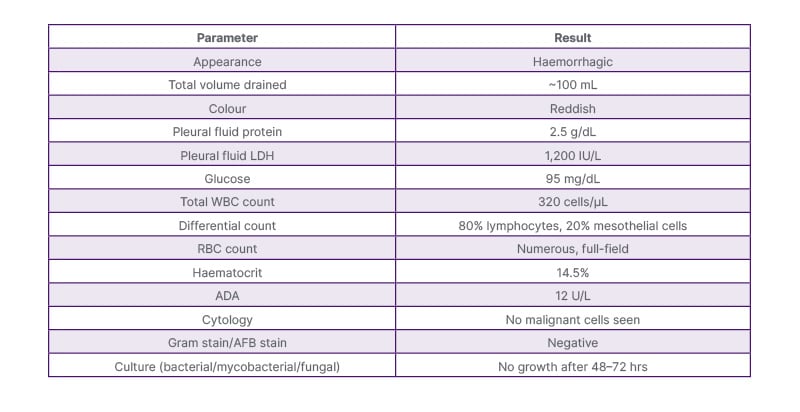
Table 1: Pleural fluid analysis report.
ADA: adenosine deaminase; AFB: acid-fast bacilli; LDH: lactate dehydrogenase; RBC: red blood cells; WBC: white blood cells.
A high-resolution CT of the thorax was done for further evaluation, which revealed a gross left-sided hydropneumothorax with complete collapse of the left lung and a 27×9 mm well-defined soft tissue density in the left breast (Figure 2). The breast lesion, confirmed as a fibroadenoma on histopathology, was considered an incidental finding unrelated to the current presentation.
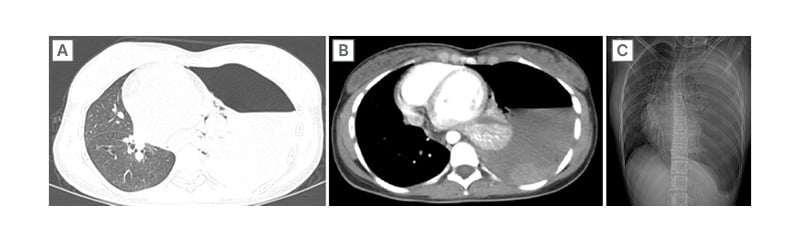
Figure 2: High-resolution CT of the thorax showing a gross left-sided hydropneumothorax with complete collapse of the left lung.
A 2D echocardiogram echocardiogram revealed mitral valve prolapse with a moderate eccentric jet of mitral regurgitation. Pleural fluid analysis was inconclusive (transudative, low adenosine deaminase). A repeat high-resolution CT of the chest showed mild residual left-sided hydropneumothorax and emphysematous bullae in the left upper lobe.
Given the constellation of clinical features, tall stature, skeletal anomalies, mitral valve prolapse, and spontaneous pneumothorax, Marfan syndrome was initially suspected. Genetic testing was done for further evaluation of the patient. Molecular analysis revealed a pathogenic FLCN gene deletion (c.1285del, p.His429ThrfsTer39), consistent with BHDS (Table 2). A heterozygous single base pair deletion in exon 11 of the FLCN gene (chr17:g.17216402del; Depth:104x) that results in a frameshift and premature truncation of the protein 39 amino acids downstream to codon 429 (p.His429ThrfsTer39; ENST00000285071.9) was detected. Subsequently, the patient underwent further testing to assess for other manifestations of BHDS. MRI of the abdomen showed no renal abnormalities. Family history was enquired; however, there was no history of any genetic disorders or similar illnesses among family members. No significant psychosocial stressors were reported. The patient is a student and lives with her family.
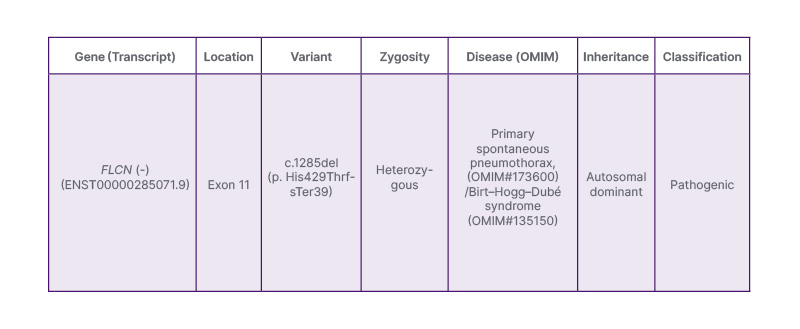
Table 2: Molecular analysis showing pathogenic FLCN gene deletion (c.1285del, p.His429ThrfsTer39), consistent with Birt–Hogg–Dubé syndrome.
Clinically, the patient showed complete resolution of symptoms, including chest pain, with full lung re-expansion confirmed by imaging. Based on clinical and radiological evidence of lung re-expansion and symptomatic improvement, the ICD was removed after 13 days (Figure 3). She was discharged in stable condition. Table 3 shows the case timeline. The patient reported marked symptomatic improvement, no discomfort post-ICD removal, and no new complaints on follow-up. No adverse or unanticipated events occurred during hospitalisation or in the post-discharge period. The patient remained stable throughout the clinical course and follow-up. The patient adhered well to all interventions. Tolerability was assessed through daily clinical evaluations and patient-reported feedback during hospitalisation and outpatient visits.
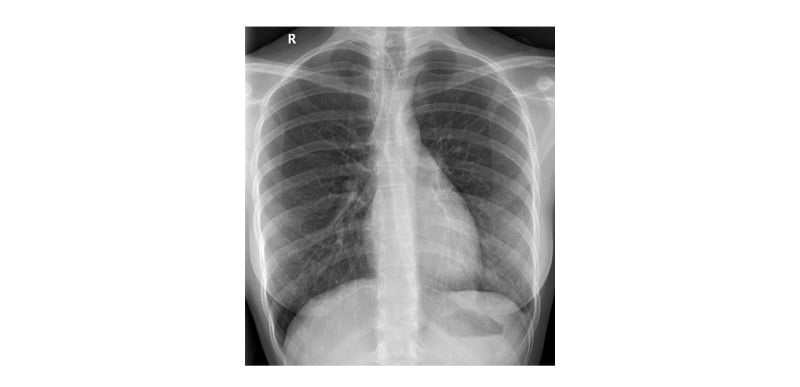
Figure 3: Chest X-ray after intercostal drain removal showing expanded left lung.
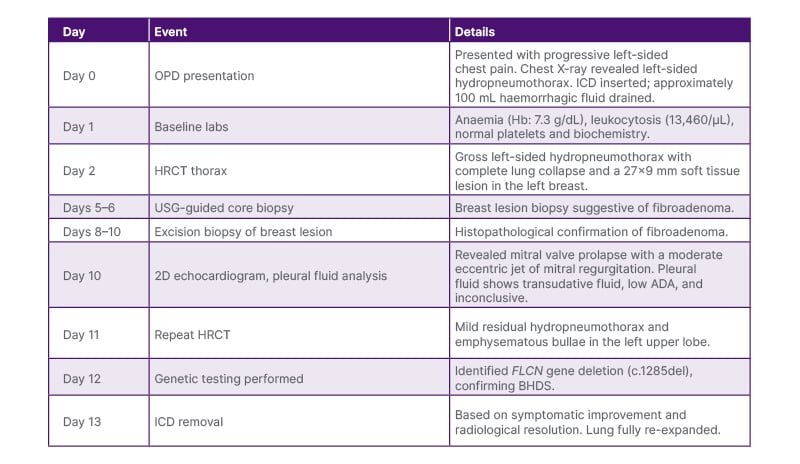
Table 3: Case timeline.
ADA: adenosine deaminase; BHDS: Birt–Hogg–Dubé syndrome; Hb: haemoglobin; HRCT: high-resolution CT; ICD: intercostal drain; OPD: outpatient department; USG: ultrasound-guided.
DISCUSSION
BHDS is an uncommon autosomal dominant disorder first described by Birt, Hogg, and Dubé in 1977, characterised by fibrofolliculomas, pulmonary cysts, spontaneous pneumothoraces, and renal neoplasms.12 The disease results from germline mutations in the FLCN gene located on chromosome 17p11.2. FLCN encodes folliculin, a tumour suppressor protein that regulates mTOR and adenosine monophosphate-activated protein kinase pathways involved in cellular metabolism, proliferation, and adhesion.6,13
Pulmonary manifestations are among the earliest and most frequent clinical features of BHDS, with thin-walled cysts seen in up to 85% of affected individuals.14 These cysts tend to be bilateral, subpleural, and predominantly located in the lower lobes and mediastinal regions, distinct from the apical pattern typically observed in primary spontaneous pneumothorax.15 Pneumothorax occurs in approximately 25–35% of patients with BHDS, often recurrent and bilateral.7 However, spontaneous haemopneumothorax, as in the authors’ case, is exceedingly rare, with only isolated reports in the literature.16,17 The haemorrhagic nature likely results from the rupture of vascularised bullae or torn pleural adhesions associated with underlying cysts.
In the present case, the patient was a 23-year-old female with no family history or cutaneous lesions, which contributed to diagnostic uncertainty. The initial suspicion of Marfan syndrome was based on her tall habitus, skeletal features, and mitral valve prolapse. Such phenotypic overlap between connective tissue disorders and BHDS has been described previously, underscoring the need for molecular testing to establish a definitive diagnosis.18
Genetic confirmation revealed a pathogenic FLCN deletion, c.1285del (p.His429ThrfsTer39), a well-documented hotspot mutation associated with BHDS across diverse populations.9,19 Frameshift variants like this lead to truncated folliculin protein and loss of tumour-suppressor function, contributing to the pleuropulmonary and renal manifestations of the syndrome. Interestingly, the authors’ patient lacked both cutaneous lesions and renal involvement, aligning with studies showing that Asian populations may exhibit a pulmonary-predominant phenotype with fewer skin and renal findings.20,21 The variable expression of BHDS reflects incomplete penetrance of FLCN mutations. Although FLCN encodes a tumour-suppressor protein involved in mTOR and adenosine monophosphate-activated protein kinase signalling, the extent of pulmonary, cutaneous, and renal involvement differs widely among individuals.7,12 Pulmonary cysts occur in up to 85–90% of patients, while spontaneous pneumothorax develops in about 25–35%, often as the first manifestation.15 Cutaneous fibrofolliculomas are absent in nearly one-third of cases, particularly in Asian cohorts, leading to under-recognition of the disorder.20 The global prevalence is estimated at roughly one in 200,000, though this is likely underestimated. In the authors’ patient, the absence of skin or renal findings despite a pathogenic FLCN mutation illustrates incomplete penetrance and the pulmonary-predominant phenotype described in Asian populations, reinforcing the need for genetic evaluation even in atypical presentations.20
The pleural fluid in this case was haemorrhagic yet biochemically transudative, likely due to the rupture of subpleural bullae with limited inflammation. Similar findings have been reported in non-infective secondary spontaneous pneumothoraces related to cystic lung diseases.22 Early drainage with intercostal tube insertion and supportive management led to full lung re-expansion and clinical recovery.
Early recognition of BHDS is vital due to its association with renal tumours, including chromophobe renal cell carcinoma and hybrid oncocytic tumours, which may occur even in the absence of cutaneous signs.23 Genetic counselling and family screening are essential, as asymptomatic carriers may present later with renal malignancy or recurrent pneumothorax.1 Current guidelines recommend baseline and periodic renal imaging (preferably MRI every 1–2 years) and surveillance for pulmonary complications.24
This case highlights several key learning points: 1) BHDS should be suspected in young, non-smoking patients with spontaneous or recurrent pneumothorax, especially when accompanied by skeletal or cardiac anomalies; 2) phenotypic variability necessitates a high index of suspicion even in the absence of skin or family history; and 3) genetic confirmation enables risk stratification and longitudinal monitoring for life-threatening renal tumours.
CONCLUSION
This case underscores the diagnostic challenges of BHDS, particularly in patients lacking family history or cutaneous signs. Spontaneous haemopneumothorax, though rare, can be a presenting feature. Early recognition through genetic testing is essential for appropriate management, surveillance of renal malignancies, and prevention of recurrent pneumothoraces.