Abstract
Necrotising enterocolitis (NEC) is a serious gastrointestinal clinical disorder primarily affecting preterm newborns. It is characterised by a wider histological spectrum, ranging from mild mucosal injury, microvascular thrombosis, and localised necrosis, to transmural intestinal necrosis. In the most severe form, it can be fatal, with complications such as intestinal perforation, peritonitis, and sepsis. Medical management is not clinically efficacious except in mild and self-limiting cases, as it is only focused on symptomatic treatment. Intestinal cell death is regarded as a crucial nascent cellular event in the pathogenesis of NEC that leads to a leaky intestinal barrier, as well as local and systemic inflammation. Therefore, cellular events that besiege intestinal cell death in NEC should be understood in a meticulous and precise manner. This review provides an extensive overview of the different types of cell death in NEC, including apoptosis, necrosis, autophagy, necroptosis, and pyroptosis. This is critically important as it helps us to comprehend the downstream signalling events that play a vital role in the initiation and progression of disease in NEC. Pertinent research studies performed in this regard would unravel novel molecular targets that could form the basis for drafting innovative therapeutic agents for optimising clinical outcomes in NEC.
Key Points
1. This article focuses on necrotising enterocolitis (NEC); a gastrointestinal disorder primarily affecting preterm newborns. Non-specific clinical presentation and rapid disease progression predispose them to develop local and systemic complications. Although most cases are managed with medical management, approximately 20–40% of cases are referred to surgical therapy. Unfortunately, surgery is associated with 50% mortality and risk of complications (strictures, abscesses, short bowel syndrome, and recurrent NEC).2. Mostly localised to the distal small intestine or proximal colon, the pathological findings range from intestinal inflammation to transmural intestinal necrosis. Intestinal cell death is regarded as a crucial nascent cellular event in the pathogenesis of NEC. The different types of cell death in NEC are reviewed in this paper, including apoptosis, necrosis, autophagy, necroptosis, and pyroptosis. It is regarded as a harbinger for a leaky intestinal barrier, intestinal complications, systemic inflammatory response syndrome, and multiorgan failure.
3. Comprehending the underlying critical pathogenic mechanisms that provoke intestinal injury, perturb intestinal homeostasis, as well as augment intestinal permeability in NEC, is a current pressing need, which this article aims to rectify. Necessary research studies implemented in this regard might unravel novel molecular targets that play a crucial role in provoking intestinal cell death in the NEC. This can form the foundation for crafting disease-specific cell-based novel therapeutic interventions for counteracting cell death, halting disease progression, and decreasing clinical mortality in NEC.
INTRODUCTION
Necrotising enterocolitis (NEC) is a gastrointestinal disease affecting preterm and very low birth weight infants (birth weight of <1,500 g, and born at <32 weeks gestation), as well as extremely low birth weight infants (birth weight of <1,000 g, and born at <28 weeks gestation).1,2 In the neonatal intensive care unit (NICU), NEC is one of the most common, costly, and critical gastrointestinal emergencies associated with increased morbidity and mortality in preterm infants. A retrospective study evaluating NEC cases from January 2014–January 2019 revealed that the overall survival of NEC infants is around 44.2%, and it is strongly associated with sex (hazard ratio: 3.10; 95% confidence interval [CI]: 1.21–7.93; p=0.018) and NEC staging (hazard ratio: 0.44; 95% CI: 0.22–0.87; p=0.019).3 Upon analysis of live births from the years 1999–2020, infant mortality due to NEC in Black, White, and mixed race infants is approximately 16.1 per 100,000 live births (95% CI: 13.1–19.2), 6.4 per 100,000 live births (95% CI: 5.5–7.4), and 10.2 per 100,000 live births (95% CI: 10.0–10.4), respectively.4
The mortality of NEC is estimated to approximately range from 10–50%.5 In a multicentre cohort study performed between 2005–2017, the mortality rate of NEC was 36.7% in 2005–2008 compared with 26.6% in 2016–2017.6 Nevertheless, in preterm infants presenting with severe complications, such as full-blown tissue necrosis, perforation, and peritonitis, the mortality is almost 100%.5 According to a 2020 report, the overall mortality of NEC is around 23.5%, with lower birth weight infants (<1,000 g) with surgical NEC experiencing relatively higher mortality (50.9%) compared with non-surgical patients.7 A 15-year outcome study from the UK that examined the NEC cases from years 2000–2015 revealed that the 30-day mortality of NEC is approximately 18.9%, and it was significantly correlated with the gestational age, area of bowel involvement, and presence of pneumoperitoneum.8
According to a recent systematic review and meta-analysis, global incidence of NEC was estimated to be around 6–7%.9 According to a retrospective follow-up study, 6.8% of the premature infants with gestational age 28–32 weeks develop NEC within 1–7 days of newborn life.10 In a Spanish study, which included 25,821 preterm infants (<32 weeks gestation) between January 2005–December 2017, the incidence of non-surgical NEC increased from 4.8% in 2005 to 6% in 2016. On the contrary, the incidence of surgical NEC demonstrated a decreasing trend (3.9% in 2005 versus 2.8% in 2016).6 In another study including low birth weight infants admitted to NICU in Pediatrix Medical Group (Sunrise, Florida, USA) from 1997–2015, non-Hispanic Black and Hispanic infants developed NEC at a higher rate (adjusted odds ratio [aOR]: 1.31; 95% CI: 1.24–1.39; p<0.001) compared with non-Hispanic White infants (aOR: 1.30; 95% CI: 1.21–1.39; p< 0.001).11 In the same study, non-Hispanic Black and Hispanic infants who developed NEC experienced higher mortality (aOR: 1.35; 95% CI: 1.15–1.58; p<0.001), compared with non-Hispanic White infants (aOR: 1.31; 95% CI: 1.09–1.56; p=0.003). The prevalence of NEC is around 1–5 in low birth weight infants.12 Analysis of 24,731 infants (<34 weeks gestational age) from May 2015–May 2018 in 25 tertiary care hospitals in China indicate incidence of NEC in very low birth weight (<1,500 g) and extremely low birth weight (<1,000 g) infants is 4.8% and 7.6%, respectively.13 Furthermore, the overall case-fatality rate and case-fatality of surgical NEC in the previous Chinese study is 9.5% and 13.6%, respectively.13
A retrospective cross-sectional study performed in Ethiopian public hospitals from 25th March– 10th May 2020 indicated that prevalence of NEC was 25.4% (n=89; 95% CI: 21.1–30.0), which is much higher compared with the USA (7.0%).2,11 Higher prevalence of NEC in Ethiopia compared with the USA can be attributed to differences in population demographics, and healthcare access, between them.2
Regardless of recent developments in the obstetric care of high-risk pregnancies, the incidence and prevalence of NEC have gradually increased globally at a steady pace over the past 25 years.2 There are no disease-specific therapeutic interventions that can reverse the clinical progression of NEC after the onset of intestinal inflammation with the pathogen insult. Even after surgical referral, the clinical prognosis of moderate to severe NEC is not very encouraging, as it is associated with many end-organ and systemic complications. NEC accounts for 2.0–7.5% of NICU admissions, and there is some variation between high-income and low-income countries.1,9,14,15 The average incidence of NEC in the USA is approximately 1.1–3.0 per 1,000 live births, and its prevalence is around 1–5% of the total number of births in the NICU.12,16,17 Race-based analysis performed in upstate New York, USA, revealed that non-Hispanic Black infants had a higher incidence of NEC compared with non-Hispanic White infants (2.2 versus 0.5 per 1,000 live births).18 Multicentre cohort studies reported a higher incidence of mortality in non-Hispanic Black and Hispanic infants compared with non-Hispanic White infants (aOR: 1.35; 95% CI: 1.15–1.58; p<0.001 and aOR: 1.31; 95% CI: 1.09–1.56; p=0.003, respectively).11 Additionally, it was reported that non-Hispanic Black infants had higher incidence of first week (aOR: 2.29; 95% CI: 2.21–2.37), neonatal (aOR: 2.23; 95% CI: 2.17-2.30), post-neonatal (aOR: 1.74; 95% CI: 1.68–1.81), and infant mortality (aOR: 2.05; 95% CI: 2.00–2.15) compared with non-Hispanic White infants.19 In the USA, the average cost of surgical procedures in NEC, which usually entails peritoneal drainage with or without laparotomy, can range from 300,000–660,000 USD.20 Despite these high costs, the clinical outcomes of surgically corrected NEC are poor, and are associated with many end-organ sequelae. The incidence of complications in advanced NEC includes short bowel syndrome (20–35%), intestinal strictures (12–35%), stoma complications (50%), neurodevelopmental impairment (30–50%), and growth delay (10%).21 It is important to note that most NEC cases occur in infants who are born at 32 weeks gestation, and with a body weight of <1,500 g.1 In clinical cohort studies, the incidence rates of NEC in infants weighing 401–750 g, 751–1,000 g, 1,001–1,250 g, and 1,251–1,500 g is approximately 11.5%, 9.0%, 6.0%, and 4.0%, respectively.16
CLINICAL PRESENTATION RISK FACTORS, PATHOLOGY, AND CURRENT TREATMENT MODALITIES
The clinical presentation of NEC usually starts with non-specific symptoms, such as rapid respiration, periodic breathing, lethargy, and irritability.21,22 Later, the preterm infant starts developing abdominal swelling; diarrhoea with bloody stool; constipation; vomiting; bradycardia; hypotension; difficulty in feeding; apnoea; lethargy; and fluctuating heart rate, blood pressure, and body temperature.21,22
In some cases, spillage of intestinal inflammation can cause pathological sequelae in the brain, resulting in neuro-inflammation, serious neurodevelopmental delay, and microcephaly.23,24 X-ray examination of the abdomen might reveal non-specific signs, such as widespread bowel distension, abdominal wall thickening, and fixation of intestinal loops until disease progression leads to intestinal perforation with peritonitis.25 Specific radiological signs diagnosing NEC can include pneumatosis, portal venous gas, pneumoperitoneum, and gasless abdomen.26 A recent study demonstrated that ultrasonography is far better and more advanced than X-ray in managing NEC, because it is more reliable in detecting bowel thickness, vascular perfusion, peristalsis, and intestinal perforation, and is associated with optimal clinical outcomes.27,28 Ultrasonography of the abdomen provides little more information, ranging from wall thickness >2.6 mm, abnormal bowel wall echoic pattern, as well as increased vascularity of bowel tissues and mesenteric peri-visceral tissues in earlier stages of NEC.25 In the later stages of NEC, ultrasonography might reveal the presence of microbubbles in a circumferential pattern within one or more loops of the intestine, the presence of air in the main portal vein or intra-hepatic portal venous branches or liver parenchyma, microbubbles appearing between the front surface of the liver and abdominal wall, and a reduction of intestinal wall thickness due to bowel ischaemia.25,29 Some of the alarming signs demonstrated by ultrasonography that necessitate immediate surgical attention include focal fluid collections, complex echogenic ascites, bowel wall thickness less than 1 mm, loss of intestinal peristalsis, increased intestinal wall echogenicity, and loss of intestinal wall perfusion.29
Other clinical signs in NEC include signs of peritonitis, abdominal tenderness, hypotension, hypovolaemic shock, metabolic acidosis, and thrombocytopenia.21 Although no specific aetiological cause has been identified so far, previous studies identified a few risk factors that can potentially make preterm infants susceptible and vulnerable to the disease. Some of the risk factors hypothesised for developing NEC include sepsis, chorioamnionitis, patent ductus arteriosus, indomethacin therapy, glucocorticoid therapy, mechanical ventilation, antenatal cocaine use, perinatal asphyxia, Black race, prolonged course of antibiotic therapy, intra-hepatic cholestatic syndrome of pregnancy, meconium aspiration syndrome, congenital heart disease, prolonged rupture of membranes, anaemia, blood transfusions, pregnancy-induced hypertension, hypoalbuminemia, formula feeding, prolonged parenteral feeding, immature intestinal motility, immature intestinal barrier, immature intestinal immunity, and abnormal bacterial colonisation.16,30-34
The underlying hallmark histological features of NEC include intestinal inflammation, enterocyte apoptosis, decreased enterocyte proliferation, mucosal oedema, haemorrhage, transmural necrosis, necroptosis, and distortion of villus and cryptic architecture. These changes are mostly localised to the distal small intestine and proximal colon.16,35,36 In a clinical research study entailing the inspection of the resected intestinal specimens of infants with NEC who underwent bowel resection over a 10-year period, it was revealed that the depth of bacterial invasion, transmural necrosis, and presence of bacteria in tissue specimens was directly proportional to increased morbidity and mortality in these preterm babies.37 In the most severe form, it has been shown to be associated with various end-organ and systemic complications, such as peritonitis, intestinal rupture, sepsis, and multi-organ failure, resulting in higher mortality in premature infants.16,38 Most of the preterm infants with NEC are managed with medical management, and approximately 20–40% of clinically severe cases are referred for surgical management. The case fatality rate of these surgically managed NEC cases is very high (50%).39
All infants with NEC are initially managed with medical therapy. The main aim of medical management is to restore intestinal homeostasis and facilitate intestinal tissue healing. Medical therapy usually entails administrating interventions, including bowel rest and tissue regeneration, gastric decompression, systemic antibiotics, and parenteral nutrition.40 Initially, mechanical ventilation and tracheal intubation might be necessary to secure the airway.41 Peripheral arterial access should be established so that arterial blood gases and systemic blood pressure can be frequently monitored.41 Signs of hypovolaemia and low blood pressure should be actively looked for so that intravenous fluids can be administered in a timely manner.41 In rare scenarios, hypoalbuminaemia due to capillary leak might necessitate the administration of colloids to combat fluid losses.41 If the infant does not respond to above-mentioned fluid resuscitation, then administrating pressors, such as epinephrine and dopamine, might be required.41 Treatment of bacterial infections associated with NEC usually includes initial delivery of a cocktail of broad-spectrum antibiotics including vancomycin, gentamycin, and clindamycin.40,41 However, due to the recent rise in the development of resistant bacterial species secondary to unrestrained antibiotics usage, their choice should be guided by incidence and prevalence of resistant bacterial cultures in the respective NICU unit.41 As NEC has been shown to be associated with medical complications, such as electrolyte disturbances, acid-base imbalance (metabolic acidosis), and coagulopathy, these should be adequately addressed by providing appropriate therapies.40,41 In addition to these therapies, frequent measurements of abdominal girth, X-ray, and abdominal sonographs should be performed regularly to monitor for any onset of local intestinal complications that warrant immediate referral to surgical therapy.41 In a recent retrospective study, it was demonstrated that medical therapy afforded lower mortality rate compared with surgical intervention (15.6% versus 50.0%).42
The infants with NEC who deteriorate in spite of medical therapy, due to rapid disease progression, should be referred for surgical management. The indications for referral to surgical management in NEC include pneumo-peritoneum, portal venous gas, ascites, fixed persistent intestinal loop, and rapid clinical deterioration.35 Surgically corrected NEC cases frequently have associated post-operative complications, such as wound dehiscence, wound infections, intra-abdominal abscesses, intestinal strictures, adhesions, cholestasis, short bowel syndrome, and recurrent NEC.16,35,43-45 Therefore, earlier detection, prompt diagnosis, and supportive management is the key to preventing morbidity and mortality in infants with NEC.
CAUSES OF EPITHELIAL CELL DEATH IN NECROTISING ENTEROCOLITIS
One of the most important causes of intestinal epithelial cell death is hypoxia.46 Xue et al.47 demonstrated increased levels of hypoxia in the intestinal tissues of ulcerative colitis and Crohn’s disease by immunohistochemistry. Some of the important reasons speculated for occurrence of hypoxia during intestinal inflammation include oedema, vasoconstriction, vasculitis, and increased O2 consumption.46 Due to the above-mentioned reasons, intestinal epithelial cells were found to be extremely hypoxic in the mouse models of intestinal inflammation.48 Moreover, bacterial infection of the intestinal epithelial cells also causes hypoxia via inciting vasculitis, as well as by increasing the O2 demand in the surrounding intestinal milieu.49 Fingerprints of hypoxia were widely demonstrated in the colitis intestine by measuring the levels of hypoxia-inducible factor, which is regarded as an important hypoxic marker.49 Intestinal inflammation tends to promote overexpression of hypoxia-inducible factor, whose main function is to preserve the integrity of intestinal epithelial barrier, and safeguard against epithelial apoptosis through nuclear transcription of protective genes.46 Hypoxia-inducible factor-1 is directly involved in regulating the ratio of T helper 17 cell/regulatory T cells in the intestinal milieu, and this dysregulation might result in increased production of proinflammatory cytokines, leading to intestinal epithelial cell death.50
Intestinal dysbiosis is defined by the downregulation of commensal bacteria and the upregulation of pathogenic bacteria resulting in the intestinal hyper-inflammatory storm.51 In a few cases, pathogenic bacterial infection might result in the destabilisation of the intestinal epithelial barrier, resulting in increased intestinal permeability, bacterial translocation, and intestinal cell death.51
Lipopolysaccharide (LPS) released from the pathogenic bacteria can act through Toll-like receptor-4 (TLR4) in the intestinal epithelial cells and neighbouring immune cells.52 LPS induced activation of TLR4 results in the secretion of cytokines (IL-6, IL-8, IL-10, and TNF-α), chemokines, and immune cell recruitment, leading to exacerbated inflammatory response and enterocyte cell death.51,52 After enterocyte cell death occurs, disrupted TLR4 signalling is hypothesised to limit tissue healing process by impairing the migration of intestinal stem cells from base of the intestinal crypt to the villus tips, thereby delaying the replacement of dead epithelial cells with fresh ones.53
Previous studies also demonstrated that increased TLR4 activation in the endothelial cells of the small intestine can impair intestinal microcirculation by downregulating nitric oxide synthase production, resulting in intestinal ischaemia and enterocyte death.54 Although neutrophil recruitment is regarded as an important first-line defence mechanism following intestinal epithelial injury in NEC, excessive neutrophil accumulation as well as their defective removal can be detrimental by contributing to intestinal epithelial cell death via release of reactive oxygen species (ROS), oxidative stress, and further release of cytokines.55 Furthermore, alteration of balance between cytotoxic (T helper 17 cell) and regulatory T cells in the intestinal milieu is postulated to be one of the most important predisposing factors for hyper-inflammatory state in the NEC intestinal tissues.56 Proinflammatory cytokines secreted by cytotoxic T cells, mainly TNF-α and interferon (IFN)-γ are the most common factors implicated in the disruption of tight junctions between the epithelial cells by altering the lipid composition, as well as by swapping the fatty acyl moieties in the phospholipids of tight junctions.57 In few instances, IL-17 secreted by cytotoxic T cells is also capable to instigate disrupted epithelial tight junctions, excess epithelial cell death, and attenuated epithelial proliferation.58 Using colon biopsy samples from patients with inflammatory bowel disease, it was recently demonstrated that IFN-γ and TNF-α were implicated in tight junction openings, resulting in intraepithelial seepage of pathogenic bacteria.59
Some previous research studies tried to investigate and identify the pathological mechanisms that underlie intestinal epithelial cell death in preterm infants with NEC. Excess ROS production, oxidative stress, and inflammation are some of the well documented mechanisms for intestinal cell death in premature infants with NEC.60,61 TNF-α, which is primarily produced by LPS stimulation, has been implicated in the intestinal epithelial cell death via the downregulation of tight junction proteins, increasing autophagy and suppression of proliferation.51 Platelet-activating factor (PAF), hypoxia-reoxygenation, ischaemia-reperfusion, peroxynitrite, bacterial invasion, and endotoxin are some other additional mechanisms proposed for the execution of intestinal cell apoptosis.3 Interestingly, the downregulation of epithelial growth factor in the serum and saliva in infants also predisposes to intestinal epithelial cell death in NEC through compromised mucosal barrier function in the intestine.62,63 Overexpression of β-arrestin-2 (cytosolic protein) can cause apoptosis of intestinal epithelial cells by pathological endoplasmic reticulum stress, which promotes the release of pro-apoptotic molecule BCL-2 interacting killer (BIK) and the activation of caspase-mediated signalling mechanism.64 The various mechanisms of intestinal cell death that occur in NEC are summarised in the next section, where each type of cell death and their relevance to the pathogenesis of the disease process are discussed.
TYPES OF CELL DEATH IN NECROTISING ENTEROCOLITIS
The different types of cell death that can occur in NEC can be broadly classified into five categories: apoptosis, necrosis, necroptosis, autophagy, and pyroptosis (Figure 1).
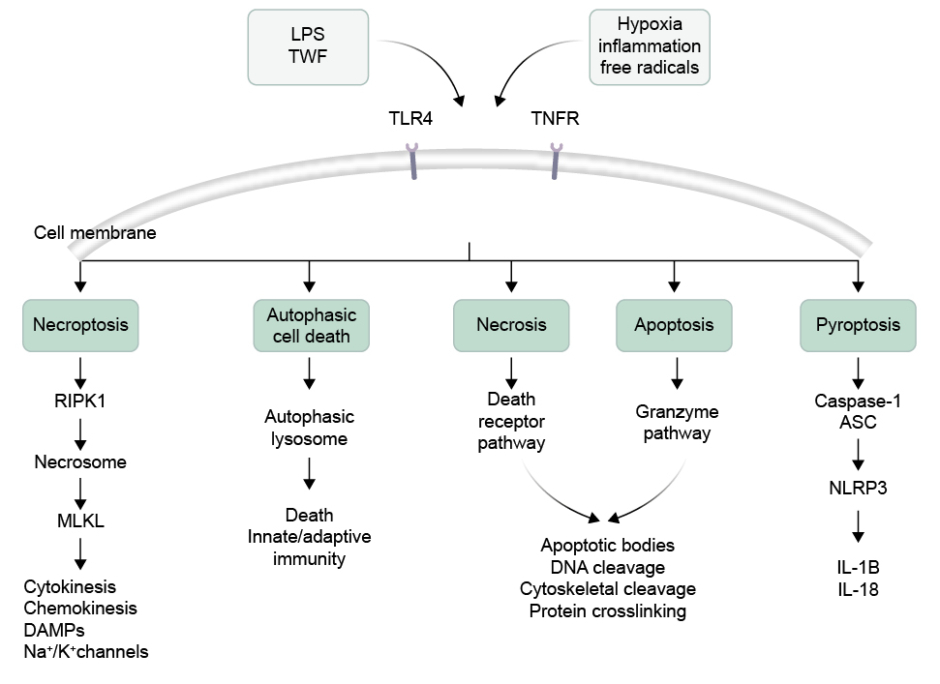
Figure 1: Illustration of different types of intestinal cell death.
Activation of TLR4 and TNFR could produce necroptosis by formation of necrosome by involving the RIPK1 and MLKL. Intestinal cells can also undergo autophagic cell death due to formation of lysosome. The necrosis and apoptotic cell death is followed by death receptor pathway and granzyme formation inducing DNA cleavage. In another pathway, caspase-1 release and NLRP3 activation produces pyroptosis.
ASC: apoptosis-associated speck-like protein containing a CARD domain; DAMP: damage-associated molecular pattern; K⁺: potassium ions; LPS: lipopolysaccharide; MLKL: mixed lineage kinase domain-like protein; Na+: sodium ions; NLRP3: nucleotide binding oligomerisation domain-like receptor family pyrin domain containing 3; RIPK1: receptor-interacting protein kinase 1; TLR4: Toll-like receptor 4; TNFR: TNF receptor.
Apoptosis
Apoptosis is one of the most important types of intestinal cell death seen in NEC tissues. Apoptotic cell death in NEC can occur in three pathways, namely the death receptor pathway (extrinsic), mitochondrial pathway (intrinsic), and perforin/granzyme pathway (T cell-mediated) pathways. All these pathways converge over the final execution pathway (caspase-3 cleavage), ultimately leading to apoptotic type of intestinal cell death in NEC. The sequelae of intestinal apoptosis in NEC can range from DNA fragmentation, degradation of cytoskeletal proteins, and cross-linking of proteins, to formation of apoptotic bodies.65
One of the most common sites for occurrence of apoptotic type cell death in NEC is the small intestine (ileum).66 Bacterial invasion and proliferation is the cardinal reason for apoptotic type of cell death in NEC.5 The intestinal micro-environment gets exposed to other pathological stimuli once bacterial infection of intestinal epithelial cells ensues. Some of these inflammatory stimuli that are implicated in the apoptotic cell death in NEC include TNF-α, LPS, ischaemia, and ischaemia-reperfusion injury.67 Accumulation of TNF-α in the intestinal cells provokes apoptotic type of cell death via Jun N-terminal kinases/p38 (mitogen-activated protein kinase [MAPK] pathway), as well as through production of ROS.68 On top of that, poor blood flow to the intestines with resultant ischaemia and immature intestinal epithelial barrier in preterm babies also increases the propensity of intestinal epithelium to undergo apoptotic cell death.69 Under the influence of these pathological stimuli, recovery and regeneration following intestinal desecration is derailed. As a result, there is excessive pathological shedding of intestinal epithelial cells leading to mechanical defect in the intestinal epithelial barrier. This will, ultimately, pave the way for superfluous bacterial invasion, massive ingress of inflammatory cytokines, extensive gut inflammation, systemic complications, and sepsis.67,70,71 In a rat model of NEC, unhindered and rapid progressive apoptosis in the intestinal epithelial cells can be a harbinger for kickstarting ensuing gross bowel necrosis of the intestinal tissues.3
In NEC, apoptosis can be considered as a defence mechanism to curb the local bacterial spread as well as to eliminate the infection. This is accomplished by sequestering and destroying the infected intestinal epithelial cells through activation of host defence mechanisms during apoptosis.72 This is important as earlier bacterial eradication will speed up intestinal repair, regeneration, and tissue homeostasis. The process of apoptosis usually occurs in acute and chronic stages. That being said, apoptosis in acute stage is protective by limiting tissue spread, whereas in the chronic stage it metamorphoses into a counterproductive process by amplifying the proinflammatory environment. So, chronic apoptosis can be considered as a harbinger for accelerated disease progression and tissue complications, thus leading to an increase in mortality and morbidity in NEC.
The process of acute apoptosis in NEC commences with bacterial proteins interacting with receptors expressed on the epithelial cell membrane (Figure 2). Bacterial LPS acts on the TLR4 receptors on the intestinal epithelium, leading to decreased proliferation and increased apoptosis of crypts and villi through involvement of p53 upregulated mediator of apoptosis.73 Upon bacterial infection, intestinal epithelial cells (IEC) recognise the pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) through Toll-like receptors (TLR), nucleotide binding oligomerisation domain (NOD)-like receptors (NLR), and upregulated proinflammatory cytokines such as TNF-α. TNF-α usually acts through TNF receptor 1 and results in formation of complex 1 (TNF receptor type 1-associated death domain) and receptor-interacting protein kinase 1 (RIPK1).74,75 This ultimately leads to increased activation of nuclear factor NF-κ-B p105-mediated survival pathways, including proinflammatory cytokine secretion, resulting in the elimination of infected IECs from the body.72,76 Increased secretion of proinflammatory cytokines contributes to accumulation of peroxy-nitrate, thus increasing the risk of apoptotic cell death in NEC.77 LPS-TLR4 interaction within the intestinal epithelium and crypts results in increased apoptosis through endoplasmic reticulum (ER) stress-mediated by protein kinase-related extracellular signal-regulated kinases [PKR]-like ER kinase, C/EBP homologous protein, and myeloid differentiation primary response gene 88 signalling molecules.78
Failure to eliminate infection due to the persistence of microbial insults leads to progression to a chronic stage. In the chronic stage, apoptosis is no longer protective but rather counterproductive, and leads to pathological consequences and disease progression (Figure 2). In the chronic stage, IECs upregulate ligands, such as Fas ligand and TNF-related apoptosis-inducing ligand, which can act via TNF receptor 1, and leads to complex II formation (Figure 2).72,79 This leads to stimulation of initiator caspases (caspase-8) and executioner caspases (caspase-3, 6, 7, and 9), ultimately resulting in the activation of nuclear factor NF-κ-B p100 pro-apoptotic pathways and inhibition of survival pathways (Figure 2).72,79-81 The end target effects of these downstream signalling events in the IECs include increased apoptosis; decreased proliferation of intestinal epithelial cells; shattered intestinal epithelial barrier; and susceptibility to several inflammatory conditions, including necrotising enterocolitis (Figure 2).72
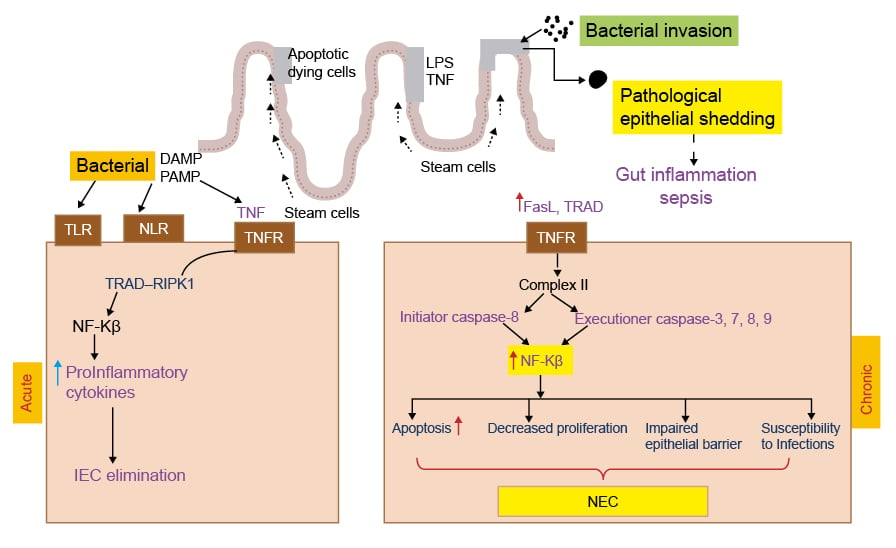
Figure 2: Representation of acute and chronic necrotising enterocolitis.
In the acute phase TLR, NLR activation enhances NF-κ-B transcription that damage IEC. Whereas in the chronic phase gut inflammation activates TNFR Complex II, which results in release of initiation and executioner caspases leading to apoptosis, decreased proliferation, intestinal epithelial barrier damage, and NEC.
DAMP: damage-associated molecular pattern; FasL: Fas ligand; IEC: intestinal epithelial cells; LPS: lipopolysaccharide; PAMP: pathogen-associated molecular pattern; NEC: necrotising enterocolitis; NLR: nucleotide binding oligomerisation domain-like receptor; RIPK1: receptor-interacting protein kinase 1; TLR: Toll-like receptor; TNFR: TNF receptor; TRAD: TNFR1-associated death domain protein.
Few studies were previously performed demonstrating the presence of intestinal apoptosis in the animal models of NEC, and by blocking apoptosis they were able to show improved clinical outcomes in these disease models. Intestinal tissues of neonatal rats exposed to formula feeding and cold/hypoxia stress had imprints of apoptosis, namely increased expression of caspase-3 and DNA fragmentation in the initial stages before progressing to transmural epithelial necrosis in the rat model of NEC.3 The LPS/TLR4/NF-κ-B signalling pathway-mediated proinflammatory cytokine secretion and mitochondrial-caspase pathway are primarily responsible for intestinal epithelial apoptosis in the rat model of necrotising enterocolitis.82 ER stress-induced activation of β-arrestin-2 causes poly-ubiquitination of binding Ig protein, and release of pro-apoptotic molecule Bcl-2 interacting killer, into the cytosol, resulting in caspase-mediated intestinal epithelial apoptosis in the animal, cell culture, and human models of NEC.83
Administering milk polar lipids decreased the intestinal apoptosis in NEC through downregulation of Bax, caspase-3 and 9 expression, as well as upregulation of anti-apoptotic Bcl-2 expression.82 Administering porcine milk exosome microRNAs resulted in decreased intestinal apoptosis and inflammation via downregulation of TLR4 pathway as well as p53 pathway in the NEC model.84 Similarly, microRNA-23a and miR-339 have been shown to decrease LPS-induced intestinal epithelial apoptosis due to their influence on TLR4 and Rho kinase inhibitor 1– sirtuin 1–NF-κ-B signalling pathways, respectively.85,86 Probiotic bacteria Lactobacillus acidophilus has been shown to attenuate intestinal epithelial apoptosis as well as cytokine production (IL-6) in the animal model and (LPS and TNF) cell culture model of NEC.87 Furthermore, another probiotic bacteria (Bifidobacterium bifidum) has been shown to attenuate intestinal epithelial apoptosis through a COX-dependent mechanism, thereby preserving intestinal mucosal integrity in the animal and cell culture models of NEC.88 Erythropoetin and epidermal growth factor have been known to have therapeutic efficacy in the animal model of NEC through reduction of intestinal epithelial apoptosis via alteration of MAPK/ERK pathway and Bax-to-Bcl-2 ratio respectively.66,89
Necrosis
Once the intestinal epithelial cells undergo apoptotic cell death, they should be cleared in a timely fashion by macrophages by a physiological process known as efferocytosis, otherwise they proceed to undergo a more devastating necrotic type of cell death.90,91 Some of the predisposing factors implicated in necrotic cell death include infectious agents, ischaemia, inflammation, and toxins.92,93 Infections are usually associated with liquefactive necrosis, which is characterised by slimy and liquefactive appearance due to the digestion of intracellular debris in dying intestinal epithelial cells by bacterial enzymes (proteases, DNases, and lysosomal enzymes).92 The morphological features of necrosis can range from cellular swelling, extensive vacuole formation, rupture of plasma membrane, and cell lysis, to nuclear and chromatin condensation in the later stages.94,95 The initial trigger for necrosis is disruption of the intestinal barrier, which recruits mucosal T-lymphocytes, and is responsible for causing upregulation of inflammatory mediators, such as TNF-α at the site of mucosal injury.94 Recruited TNF-α acts on the intestinal epithelial cells via TNF receptors to induce the expression of ROS production, which acts as a secondary messenger to incite various cellular derangements ranging from DNA damage and proinflammatory signalling pathways to membrane damage in the presence of pathological stimuli and stressors.94 Since necrosis is associated with cellular rupture, it can cause the dumping of cellular debris into the extracellular microenvironment with the inherent risk of developing secondary systemic inflammatory response syndrome.93
NEC is the most common cause of intestinal necrosis in premature neonates, and is associated with infection-related intestinal ischaemia.96 It is important to understand that bacterial invasion of the intestinal epithelium is one of the most important primary events for the occurrence of necrotic type of cell death in NEC. Hypoxia, anaerobic metabolism, swelling, rupture of IECs, and breaching the intestinal epithelial barrier are the initial events that can accentuate bacterial infiltration into the intestinal mucosa in NEC.97 Bacterial invasion of the intestinal wall can result in proinflammatory mediator secretion, as well as activation of TLR4 receptors in the mucosal microvasculature through nitrate-nitrite signalling, resulting in intestinal ischaemia and transmural necrosis.54,97
One of the most promising screening tools for monitoring the extent of transmural necrosis in the small intestine in patients with NEC is Doppler ultrasonography.98 Efforts need to be made to identify the NEC in earlier stages, before the occurrence of intestinal transmural coagulative necrosis, since its occurrence heralds the onset of severe complications, such as perforation, bacterial overgrowth, pneumatosis intestinalis, inflammation, and peritonitis.99 The prognosis of patients with above-mentioned complications is very poor and mostly necessitates surgical interventions with associated morbidity and mortality.
Intestinal epithelial apoptosis is the initial cellular event that slowly progresses to morphological transmural necrosis of the small intestine in neonatal rats subjected to formula feeding and asphyxia stress in the experimental model of NEC.3 Previous studies have demonstrated that there is no positive relationship between mortality of surgical NEC cases and severity of histological necrosis evident in the small intestine, although upregulation of immunological markers, such as cleaved caspase-3 and vascular cell adhesion protein, has been associated with poor survival.100 In another multicentre study, Heida et al.101 had demonstrated that intestinal fatty acid binding protein levels in the plasma and urine directly correlate with the extent of surgical resection in the NEC, and it has the potential to become a reliable serum biomarker for monitoring transmural necrosis of the small intestine in later stages of NEC.
Apart from bacteria and inflammatory cytokines, another important mediator that can cause bowel necrosis in NEC is platelet activating factor (PAF).102-104 A previous study reported that a combination of LPS with PAF administered intra-aortically is sufficient to induce ischaemic intestinal necrosis in rats with lesions exhibiting morphological similarity to the tell-tale signs of NEC.102 Furthermore, the Sun et al.104 study revealed that TNF-α-induced bowel necrosis is mediated by PAF as it was attenuated by administration of PAF inhibitor. In the same study, it was also demonstrated that additive synergism between LPS, TNF-α, and PAF is necessary for the occurrence of intestinal necrosis, and this was associated with hypovolaemic shock in rats treated with these toxins.104 The underlying mechanism through which PAF can induce bowel necrosis in NEC is via activation of the lipoxygenase pathway of arachnoid metabolism and upregulation of leukotrienes.103
Necroptosis
Necroptosis is a programmed type of cell death that has the features of apoptosis and necrosis, and occurs with inflammatory conditions.105 Receptor interacting protein kinase (RIPK) is a protein kinase that also fulfils a physiological role of protecting IECs from TNF induced caspase-8 apoptosis, and thereby maintaining intestinal homeostasis.106 Caspase-8 inhibits the RIPK1 and RIPK3 in the physiological conditions and prevents their interaction (Figure 3). Classical necroptosis is usually initiated by formation of complex of RIPK1 and RIPK3 when caspase-8 is inhibited, resulting in the formation of necrosome complex (Figure 3).107,108 Once the necrosome complex forms, it activates mixed lineage kinase domain-like protein (MLKL), resulting in its phosphorylation and oligomerisation. MLKL is regarded as a chief executioner of necroptosis type of cell death (Figure 3).107,108 Activated MLKL migrates to the plasma membrane and executes necroptosis by pore formation, thereby secreting cytokines, chemokines, and DAMPs, as well as engaging sodium and potassium channels (Figure 3).107
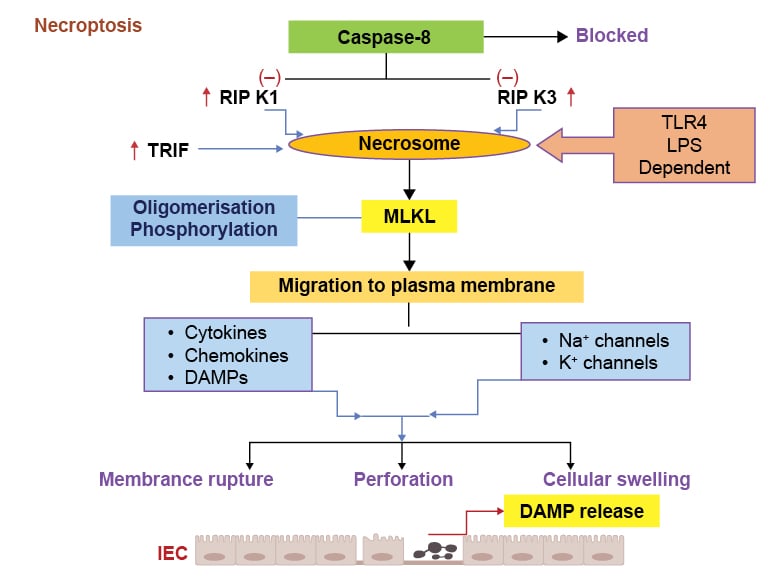
Figure 3: Various pathological events in necroptosis process.
Activation of TLR4 induces necrosome formation due to increase in the RIPK1/3 and TRIF signalling, leading to the caspase-8 inhibition. The necrosome formation causes MLKL phosphorylation and migration to the plasma membrane. These pathogenic responses induce cytokines, chemokines, and DAMPs release, which can activate Na+ and K+ channels and increase membrane permeability, rupturing, and cell swelling.
DAMP: damage-associated molecular pattern; IEC: intestinal epithelial cell; K+: potassium ions; LPS: lipopolysaccharide; MLKL: mixed lineage kinase domain-like protein; Na+: sodium ions; RIPK1: receptor-interacting protein kinase 1; RIPK3: receptor-interacting protein kinase 3; TLR4: Toll-like receptor 4; TRIF: IR-domain-containing adapter-inducing interferon-β.
The morphological features of necroptosis type of cell death include cellular swelling, membrane rupture, the release of DAMPs, and tissue inflammation.108 During bacterial infections, LPS induced activation of TLR4 receptors in the mouse macrophages initiates the formation of a complex between RIPK3 and Toll/IL-1 receptor domain-containing adapter inducing IFN-β (TRIF), resulting in the occurrence of non-classical necroptosis.109,110 Salmonella enterica typhimurium-induced interferon type I expression in the macrophages leads to RIPK1-RIPK3 induced necroptosis, resulting in macrophage depletion and evading of innate immune response.111 Alternatively, the LPS/TLR4/TRIF/receptor-interacting protein kinase-3 (RIPK3) axis in the macrophages can potentially induce the formation of NLR family pyrin domain containing 3 (NLRP3) inflammasome and caspase-1 activation through alternate pathways, resulting in the production of proinflammatory cytokines, such as IL-1β and IL-18 for the amplification of inflammatory response.112
According to Sinclair et al.,113 the development dependent enterocyte metabolism might predispose to the development of necroptosis induced intestinal injury in the NEC.113 Upregulation of necroptosis proteins (receptor-interacting protein kinase-1 [RIPK1], RIPK3, and MLKL) and activation of necroptosis occurred in Toll-like receptor dependent manner in the differentiated intestinal epithelium of immature ileum with LPS treatment of wild-type mice, as well as in a novel NEC-in-a-dish cell culture system.114 Breast milk and pharmacological inhibition of necroptosis resulted in decreased epithelial cell death and mucosal injury in the mouse model of NEC.114 In consensus with this study, Liu et al.115 demonstrated that necroptosis was induced in the patients with NEC and cell lines through the TLR4/TRIF/RIKI/MLKL axis, as well as TNF-α/TNFR/RIKI/MLKL axis.
With necroptosis proven to be involved in the pathogenesis of NEC models, efforts to inhibit necroptosis yielded encouraging results in the cell culture and animal models of NEC. Necrostatin-1 (receptor-interacting protein kinase 1 antagonist) treatment resulted in decreased necroptosis protein expression, decreased intestinal cell death, and preserved intestinal barrier in the mice model of NEC.115 In another study, LPS challenged weaning piglets treated with medium chain triglyceride diet displayed decreased expression of necroptosis proteins (receptor-interacting protein kinase 1 and MLKL) as well as TLR4 and nucleotide binding oligomerisation domain proteins, leading to enhanced intestinal barrier function, disaccharidase activities, and better intestinal morphology.116 MiR-143-3p was downregulated in the serum of patients with NEC compared with healthy controls, and its depletion promoted RIKI induced necroptosis in the LPS treated caco-2 cells.117 In the in vitro studies, miR-143-3p supplementation resulted in the decreased expression of necroptosis proteins (RIKI and MLKL), attenuated the necroptosis, and associated inflammation in the LPS-treated caco-2 intestinal epithelial cells.117
Autophagy
Autophagy (eating of self) is a self-destructive process of removal of damaged organelles, misfolded proteins, and intracellular organisms.118 There are three main types of autophagy, namely, macro-autophagy, micro-autophagy, and chaperone-mediated autophagy.118,119 The stages in autophagy include initiation, nucleation, elongation, maturation, lysosomal fusion, and degration.119 The core machinery required for the autophagic process is summarised as Initiation (ULK1 complex [FIP200, ATGB, ATG10]), nucleation (Class III phosphatidylinositol 3-kinase complex [BECWI, AMBRA, ATG14L, UPS15, UPS34]), elongation (WIPI proteins), maturation (E3 complex [ATG5-ATG12-ATG164]), lysosomal fusion (phosphatidylethanolamine [PE]), and degradation (LC3).119,120
In normal homeostatic conditions, the autophagic process is involved in the regulation of numerous physiological functions in the human intestine, including preserving barrier integrity, prevention of bacterial dissemination, host-microbial interaction, goblet cells mediated mucin secretion, macrophage-induced pathogen clearance, dendritic cell-mediated antigen presentation, effective and memory T cell development, and secondary antibody response.121 IECs infected with vibrio cholera secreting pore-forming toxin (cytolysin) responded initially with increased Microtubule-associated proteins 1A/1B light chain 3B (LC3B-II) autophagic vacuole formation to neutralise the pathogen spread.122 Next, when autophagy is inhibited in the IECs infected with vibrio cholera, they demonstrated decreased survival, which supports the presumption that autophagy is a cellular defence mechanism to localise, subdue, and destroy the intracellular bacteria.122 In another research study, as bacteria such as S. typhimurium enters the cells through entry vacuoles, they are immediately ubiquitinated, and coated with p62 so that destruction of bacteria can proceed by cellular autophagy apparatus to restrict their rapid replication and limit cell-to- cell spread.123
It is important to keep in mind that the process of autophagy not only serves to destroy the pathogen but also promotes the initiation of immune response for its destabilisation and impairment of its multiplication, and resultant cellular spread. Degradation of bacterial/viral components by autophagic process drives the activation of innate and adaptive immunity, resulting in the IFN-γ production and major histocompatibility complex Class II-mediated CD4+ T cell response, respectively.124 But, some microbial agents cleverly have intrinsic mechanisms set in place that tend to cripple the process of autophagy, and thereby escape the host immunity by impaired trafficking into the autophagolysosome and destabilising the autophagy proteins.124 For example, Shigella flexneri secretes IcsB protein due to which it escapes from binding to the autophagy apparatus (autophagy related [Atg] 5), leading to successful evading of autophagy process, and this results in its eventual multiplication and enhanced cellular spread.125
Understanding the shifts in autophagy regulation in the different stages of pregnancy is essential as it can have direct influence on the occurrence of infection in the maternal-fetal unit. It has been demonstrated that expression of autophagy marker LC3B-II in the placenta decreases during first trimester and increases during term pregnancy with TNF-α treatment.126 This fluctuation of LC3B-II during different stages of pregnancy mediated by inflammatory cytokines indicates that dysregulation of autophagy can have potential implications on the regulation of infection and apoptosis in the maternal-fetal unit.126 Autophagy is deemed to be a protective mechanism against intestinal inflammation; however, it can be counterproductive in some instances depending upon on the presence of toxins and cytokines in the intestinal milieu.
In preterm infants, increased autophagic protein gene expression and TLR4 signalling can kickstart the autophagy process that can secondarily impair intestinal epithelial migration through inactivation of Rho-GTPase, a predisposing factor that can ultimately lead to NEC.127 It is quite plausible that an increased autophagic process tends to be protective at least in the earlier phases of NEC intestinal injury, by limiting the progression of the disease.128 Electron microscopy evaluation of intestinal epithelial cells revealed that there is upregulated expression of autophagy proteins (light chain 3 and beclin), increased formation of autophagic vacuoles, as well as degradation of p62 in the ileum of neonatal rats with NEC.128
During pathogen-mediated intestinal inflammation, signalling pathways that tend to inactivate the autophagic process favour disease progression in NEC (Figure 3). Studies have shown that LPS mediates an increase in the ROS, including peroxy-nitrate production, which can lead to epithelial injury and intestinal damage in the NEC disease models.129 The increase in ROS production in the intestinal tissues also evokes inactivation of autophagy marker Atg4 and lipidation of Atg8, which in combination cripples the autophagy process in the intestinal epithelial cells. It is important to keep in mind that the decreased autophagy process in the IECs is a liability factor for development of NEC, as disease progression moves forward unchecked without this important protective mechanism.130 But in the later stages of the inflammatory disease process, the presence of continuous stressors and chronic tissue injury becomes detrimental as they convert the protective autophagic process into a destructive mechanism that is incapable of protecting against disease progression. In other words, autophagy tends to be dysregulated, counterproductive, and becomes less beneficial in controlling the infection and therefore accentuates the mucosal tissue injury in the NEC.128
Accordingly, controlling the dysregulated autophagy was used as a therapeutic strategy in regulating apoptosis and reducing tissue injury in the NEC models. In a recent study, TNF-α treatment resulted in decreased proliferation and increased apoptosis in the IECs (IEC-6 cells) through autophagy induction (increased LC3II/I ratio, beclin induction, and increased autophagy vacuoles), and use of autophagy inhibitor (wortmannin) reversed these effects, emphasising the importance of autophagy in regulation of apoptosis in the in vitro NEC models.131
In the rat model of NEC, it was revealed that autophagy preceded apoptosis and administration of erythropoietin resulted in suppressing autophagy markers (beclin 1 and LC3II), as well of apoptosis through phosphoinositide 3 kinase (PI3K)/Akt/mammalian (or mechanistic) target of rapamycin signalling pathway and the MAPK/ERK pathway, respectively.89 By controlling excessive autophagic activity in enterocytes, human β-defensin-3 (cationic host defence peptide) is able to promote epithelial migration, attenuate mucosal inflammation, maintain mucosal integrity, and thereby offer protection against mucosal injury in the neonatal rat model of NEC.132 Likewise, supplementation of epidermal growth factor was beneficial in attenuating the intestinal injury in NEC via downregulating autophagy in the in vitro and in vivo models of NEC.128
BACTERIAL INFECTION AND INNATE IMMUNE RECEPTOR RECOGNITION
Bacterial infection of IECs leads to eliciting of immune response via downstream signalling of pathogen recognition receptors (PRR).133 There different kinds of PRRs include TLRs, NLRs, retinoic acid-inducible gene-I-like receptors , C-type lectin receptors, and AIM2-like receptors.133 Lipoproteins and proteins of bacteria are recognised by TLR receptors (1, 2, 4, 5, 6, and 10), whereas nucleic acid of bacteria was identified by TLR receptors (3, 7, 8, and 9), expressed on the surface of IECs.134 These will lead to stimulation of TRIF3 (Toll/IL-1R domain containing adaptor protein inducing IFN-β) and myeloid differentiation primary response gene 88 pathways leading to activation of NF-κ-B, IFN regulatory factor 3, and MAPK (mitogen-activated protein kinase) mediated downstream signalling events.133,135 This will eventually result in nuclear transcription of proinflammatory pathways, and elicit a robust immune response against the pathogen.133,135
Sometimes, bacteria craft mechanisms to escape binding to these surface PRRs and undergo phagocytosis to enter IECs. Once inside, they will be able to bind to intracellular PRRs, such as NLRs. These intracellular NLRs have ligand recognition receptors at the C-terminal domain, central NACHT domain (a domain coined due to the presence of four NLR members, namely, NAIP, CIITA, HETE, and TP1) and caspase activation and recruitment domain or pyrin domain at the C-terminal domain.133,136-138 In physiological conditions, these NLRs are in inactive state due to their inherent U-shaped configuration, where the NACHT domain is masked and prevented from undergoing oligomerisation. As soon as bacteria are broken down by intracellular lysosomal proteases, PAMPs are released and bind to LRR. Binding of PAMPs to LRR results in the unmasking of the NACHT domain, and this leads to its subsequent oligomerisation and activation. Gram-positive and gram-negative peptidoglycans can activate NOD-containing protein 1 (and NOD-containing protein 2 receptors, leading to the stimulation of NF-κB pathways, which can be a starting point for activation of NOD leucine rich repeat pyrin domain containing protein 3 (NLRP3) inflammasome .135 Activation of NLRP3 inflammasome intracellularly promotes innate immune response by secretion of proinflammatory cytokines, and provokes a specific type of cell death called pyroptosis, which enables clearance of pathogen infected intestinal epithelial cell.139,140 It is interesting to know that some pathogens can prevent NLRP3 activation and occurrence of pyroptosis by devising various mechanisms, such as masking of ligand expression, structural alteration of ligands limiting the detection of NLRs, and direct inhibition of inflammasome.140
Pyroptosis
Bacterial infection of IECs can release DAMPs and PAMPs, which can act on NLRs.93,141 The activation of NLRP3 inflammasome occurs in a two-phased manner: NF-κ-B meditated transcription of inflammasome components in Phase I, as well as assembly and activation of NLRP3 inflammasome in Phase II.142 Activation of NLR on IECs can induce the formation of inflammasome complex comprising of apoptosis-associated speck-like protein containing a caspase activation and recruitment domain, NLRP3, and capase-1 resulting in secretion of proinflammatory cytokines, such as IL-1β, and IL-18 in the classic canonical pathway.141,143 It is important to note that IECs secrete relatively less IL-1β secretion in contrast to lamina propria macrophages, which are the main source of IL-1β during intestinal inflammation.144 Alternatively, LPS released from gram-negative bacteria can cause activation of caspase-11, resulting in cytoplasmic swelling and release of DAMPs in the non-canonical pathway.93,142 Both canonical and non-canonical pathway result in pyroptosis, a special from of cell death specific to NLRP3 inflammasome activation.93,142,143 An important distinguishing feature of the non-canonical pathway is that it can directly induce pyroptosis by stimulating pore forming protein gasdermin D, and reinforce the activation of the classical canonical pathway, thereby potentiating the secretion of pro-inflammatory cytokines.145
The main physiological function of pyroptosis is to disrupt microbial replication and make them susceptible for phagocytosis by immune cells.140
The important morphological features of pyroptosis cell death include cellular swelling, osmotic lysis, exocytosis of inflammatory intracellular contents, DNA cleavage, nuclear condensation, and disruption of actin cytoskeleton.143 The benefits of inflammasome activation in the IECs can range from proinflammatory cytokine secretion, enhanced lipid production (membrane repair), preventing autophagic cell death, recruitment of macrophages, activation of B and T lymphocytes, stimulation of dendritic cells, and wound healing, to enhanced mucosal integrity.143,144 NLRP3 inflammasome activation can be regarded as an important intestinal immune defence mechanism as it inhibits bacterial growth, promotes cellular repair, and enhances cellular survival in the earlier stages of infection.143 However, in the later stages of infection (14–21 days post infection), cytosolic NLR Type 4 (NLRC4) has been implicated in the execution of an innate immune defence mechanism against Citrobacter rodentium by attenuating bacterial colonisation and associated intestinal inflammation.146 NLRC4 inflammasome is activated by ROS, lysosomal disruption, and calcium signalling, whereas the bacterial flagellins and Type III secretion system are capable of activating NLRP4 inflammasome.147 During physiological conditions, NLRP4 complexes with beclin-1 to negatively regulate the autophagy process.148 On the contrary, during bacterial infections, it dissociates from beclin-1 to migrate towards the phagosomes containing Streptococcus A, thereby promoting beclin-1-mediated autophagic degradation of bacterium.148
In the different phases of bacterial infection, IECs activate different types of inflammasomes as a part of an innate defence mechanism to neutralise the microbial threat.146 IECs initially sample the level of bacterial load and decide to move forward with inflammasome-mediated pyroptosis, if they sense that bacterial threat is high enough to warrant the activation of the immune system for limiting the intestinal damage.142
Suppression of NLRP3 inflammasome had offered clinical benefit in ulcerative colitis and mucosal colitis, underscoring its importance in the pathogenesis of inflammatory bowel diseases.149,150 Cronobacter sakazakii causes TLR4/NF-κ-B-mediated NLRP3 inflammasome activation, leading to IL-1β secretion and pyroptosis in the in vitro and rat models of NEC.151 According to Hu et al.152 there is increased expression of IL-1β, IL-18 (mRNA and protein), and NLRP3 (mRNA) in the NEC group of rats treated with hypoxia and cold stimulation, compared with the control rats, emphasising the importance of NLRP3 inflammasome and pyroptosis in the disease pathogenesis.152
NLRP3 inflammasome has been shown to be activated in the intestine, and brain NEC models, and its blockade with inhibitor MCC950 resulted in the substantial reduction in intestinal and brain inflammation, pathological severity, as well as long term neurological sequelae.153 In the LPS-treated IECs, there was increased expression of high-mobility group protein 1 (HMGB1), NLRP3, TLR4, NF-κ-B, caspase-1, IL-1β, and TNF-α, as well as knockdown of HMGB1, resulting in the attenuation of these markers, highlighting the role of HMGB1 in inducing TLR4-induced activation of NLRP3 inflammasome.154 In line with these findings, increased expression of HMGB1, NLRP3, TLR4, NF-κ-B, caspase-1, IL-1β, and TNF-α noted in the rat model of NEC compared with the control group was reversed with treatment of HMGB1 inhibitor.154 Fork 03A protein (FOXO3a), a member of the fork protein family and a transcription factor that functions to limit the translocation of NF-κ-B into the nucleus, derailed the proinflammatory cytokine production and associated inflammasome-induced inflammation in the animal models of NEC.155 In parallel to the above findings, Yin et al.155 demonstrated lower expression of FOXO3a and higher expression of NLRP3, TLR4, caspase-1, IL-1β, and IL-18 in the intestinal tissues of NEC rats and LPS-treated IECs. Overexpression of FOXO3a resulted in downregulation of NLRP3 inflammasome cytokines and reduction of pyroptosis, thereby highlighting its role as a potential therapeutic strategy in regulating inflammasome mediated intestinal injury in the in vitro and animal models of NEC.155
MiRNA-146a-5p was shown to downregulate NLRP3 related downstream signalling molecules including caspase-1, and its upstream chloride intracellular channel 4, in the THP-1 cells treated with low/moderate doses of LPS (0.5 μg/mL and 1.0 μg/mL) and its overexpression resulted in improved survival in the animal models of NEC.156 Increased expression of suppressor of cytokine signalling 3 protein resulted in the decreased expression of NLRP3, caspase-1, IL-1β, IL-18, and TNF-α in the LPS treated caco-2 and fetal human cells, as well as intestinal tissues of NEC, highlighting its therapeutic efficacy in suppressing NLRP3 inflammasome-induced inflammation in the NEC models.157
MANY UNANSWERED QUESTIONS AND UNMET NEEDS: CONCLUSIONS AND FUTURE PERSPECTIVES
The different types of intestinal cell death that can possibly occur in NEC include apoptosis, necrosis, necroptosis, pyroptosis, and autophagy. The type and severity of cell death is mostly dependent on the various microbial insults such as type of bacteria, amount, and type of toxins released and immune responses that are present in the intestinal milieu. The current treatment strategy utilised in the treatment of NEC includes bowel rest, intravenous antibiotics, and regular radiographic monitoring.127 Despite the best efforts of clinicians in managing these NEC cases, therapeutic options currently employed have limited efficacy, and afforded just symptomatic relief while awaiting natural disease recovery with no effect in reversing the disease progression after its onset.
Therefore, it is important to comprehend and analyse the underlying critical pathogenic mechanisms that provoke the intestinal injury, perturb intestinal homeostasis, as well as augment intestinal permeability in NEC. In this regard, pathogens that invade and precipitate IEC apoptosis can compromise the intestinal epithelial barrier, and trigger a wide variety of untoward pathological consequences, such as increased intestinal permeability, intestinal inflammation, systemic inflammatory response, translocation of bacteria into the systemic circulation, systemic inflammatory response syndrome, and multiorgan failure.27 Therefore, cell death of IECs is regarded as a critical nascent cellular event in the pathogenesis of intestinal inflammatory diseases such as NEC. So, underlying pathophysiology and associated cellular signalling events surrounding intestinal cell death in the NEC need to be delved into and understood in a systematic and methodical manner.
In this review, the authors provided a detailed overview of different types of cell death in NEC with a special focus on their individual pathogenic mechanisms and signalling pathways. Discussing the pathophysiological mechanisms for various types of cell death in the NEC will be helpful for understanding the downstream signalling events based on the type of risk factors present depending on the bacterial/viral insults encountered in the intestinal milieu. Probing into the systematic downstream signalling events and mechanisms that underlie various types of IEC death can be very useful in comprehending the pathogenetic mechanisms that initiate and perpetuate disease progression in NEC. Appropriate basic science research studies implemented in this regard might unravel novel molecular targets, including membrane receptors, tyrosine kinases, and intermediate signalling factors that play a crucial role in provoking intestinal cell death, and inciting leaky intestinal barrier in NEC. The authors surmise that blocking these earlier signalling events that occur after bacterial insult with the help of these deciphered molecular targets might offer an opportunity to reverse the disease progression from the initial stages of NEC. Therefore, these molecular targets can form the structural basis for crafting disease-specific cell-based novel therapeutic interventions for counteracting cell death, halting the disease progression, and decreasing clinical severity and mortality in infants with NEC. Finally, clinical research studies to test the efficacies of these novel therapies on disease severity and systemic complications in NEC would be the logical next step and the need of the hour so that they can be implemented in the clinic for optimising clinical outcomes, as well as decreasing morbidity and mortality in NEC.