
Abstract
The purpose of this review is to provide the haematologist with a working knowledge of the common inherited bone marrow failure syndromes (iBMFS) diagnosed in early childhood to young adulthood. Although these disorders are heterogeneous, this article discusses their common features, pathophysiology, and management. Each of these syndromes has a spectrum of clinical variation and can cause both haematological and non-haematological manifestations. Most pathogenic mutations responsible are in genes important to a progenitor cell’s ability to maintain genomic integrity, which accounts for the clinical phenotypes often affecting multiple tissues. Furthermore, all of these syndromes predispose not only to aplastic anaemia but also to myelodysplastic syndrome/acute myeloid leukaemia. Since iBMFS only account for a small percentage of childhood leukaemia cases, it is important that the clinician maintains a high clinical suspicion as appropriate diagnosis impacts treatment, health screening, and family members. Identification of iBMFS is critically important for appropriate donor selection and transplant regimens, as haematopoietic stem cell transplantation is curative for the haematological manifestations of these diseases, but treatment-related mortality can be excessive if modifications are not made to conditioning.
INTRODUCTION
Inherited bone marrow failure syndromes (iBMFS) are a heterogeneous group, with variable phenotypes, inheritance, and gene mutations, but also with striking similarities. All iBMFS include failure of adequate blood cell production, and whilst some are lineage specific, most lead to progressive pancytopenia. Many of the iBMFS are associated with a decreased number of CD34+ haematopoietic progenitor cells (HPC), along with stress erythropoiesis, and a predisposition to myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML). iBMFS are often characterised by multiple non-haematological findings, such as congenital malformations and predisposition to solid tumours. Variability exists and even the same mutation in individuals within one family can vary from severe phenotypic findings to being clinically silent. Although the mutated genes vary between syndromes, most are key to cellular homeostasis and vital to dividing progenitor cells.1
When Dr Fanconi published a landmark paper in 1927 describing families with severe bone marrow failure (BMF) in conjunction with multiple congenital anomalies, it was the clinical phenotype that elucidated the diagnosis of Fanconi anaemia (FA).2 In the modern era, with improved diagnostic tools, such as clastogenic assays and gene panel sequencing, it is now apparent that FA and other iBMFS also often occur in patients with less severe clinical presentations. Accordingly, the prevalence of iBMFS is likely to be higher than the estimate of 65 per million.3,4 Today, we recognise that diagnosis of iBMFS is not just made in young children with multiple congenital anomalies and that for patients with subtler physical findings the diagnosis can be delayed to adolescence/young adulthood (AYA) during the evaluation of newly diagnosed aplastic anaemia (AA) or MDS/AML. In a study of patients who had a haematopoietic stem cell transplant (HSCT) for AA or MDS, germline mutations diagnostic of a iBMFS were identified in 5% and 14% of patients, respectively, and neither family/personal history or physical stigmata were predictive.5 Physical anomalies were present in less than a third of the patients. The peril of under-diagnosis is not only that individuals have specific health risks, but if they remain undiagnosed at the time of MDS/AML presentation they are at risk from severe toxicity, if ‘standard’ chemotherapy regimens are not adjusted appropriately.6-8 Moreover, although we now routinely test for many of the somatic mutations and cytogenetic abnormalities to inform risk-adjusted treatments during the evaluation of AML/MDS, a comprehensive evaluation of genes associated with iBMFS is not routine. Thus, recognising when to evaluate further for possible hereditary predispositions remains of great importance.3,9
FANCONI ANAEMIA
In addition to a young child with pancytopenia, well-described characteristics suggesting FA include hypoplastic thumb, radial ray skeletal defects, café-au-lait spots, microcephaly, and undescended testicles. Other non-haematological manifestations include short stature, characteristic facial findings, skeletal, renal, gut, gonadal, urogenital, cardiopulmonary, and central nervous system anomalies, as well as neurocognitive delay. Such findings direct the haematologist to order a chromosomal fragility test in the presence of DNA crosslinking agents, diepoxybutane or mitomycin C; the gold standard for FA diagnosis. Occasional false positives may occur in patients with other chromosomal breakage syndromes, such as Nijmegen breakage syndrome. False negative results on blood analysis may occur due to genetic reversion of the HPC, wherein spontaneous resolution of pancytopenia can occur through the repair of a mutated allele.10 In the setting of a high suspicion for FA, a germline mutation assay on skin fibroblasts should be performed. Identification of pathogenic FA mutations on genetic analysis is confirmatory. Clinical suspicion is important due to variable penetrance and expressivity during evaluation of an AYA with AA or MDS/AML. Approximately 10% of FA patients will not be diagnosed until they are 16 years of age or older.11 Congenital anomalies are absent in ˜40% of patients. Therefore, part of the diagnostic workup for AML or MDS should include a detailed family/personal history and careful physical exam to look for specific findings. It is easy to overlook manifestations, such as short stature, since a patient may fall within the normal range unless one calculated the patients’ predicted height based on mid-parental height. Diagnosis of FA is critically important in patients with AA or MDS/AML, because the hallmark of the syndrome is genomic instability in the presence of DNA crosslinking agents and treatment plans for these diseases typically include such agents as part of a HSCT. Furthermore, dose attenuation is required to decrease treatment-related mortality (TRM) and morbidity.12 Lastly, diagnosis allows for screening of other FA-associated complications, and is important for family planning.
FA is primarily of autosomal recessive inheritance, although there are autosomal dominant (all de novo) and X-linked inheritance as well. Currently, 21 genes have been identified that, when mutated, cause FA,13 with biallelic FANCA mutations the most common genetic aetiology. These genes are critical for the cells to repair damage caused by DNA interstrand crosslinks normally by homologous recombination. Mutations in the FA complex increase the alternate repair pathway (non-homologous recombination), which is more error prone, leading to genomic instability.1,13,14 Since in FA DNA damage is mediated by exposure, the fact that the HPC is exposed to endogenous toxins, such as oxidative stress and aldehydes, also makes it particularly sensitive.15 The DNA damage in these dividing HPC activate p53 and like pathways that lead to cell cycle arrest and either apoptosis or senescence to prevent propagation of mutations in the daughter cells, but this causes an accelerated attrition rate of HPC. In fact, FA patients have low numbers of CD34+ HPC.16 This is similar to ageing, but here the blood ages at an accelerated rate. A vicious cycle occurs of the marrow in FA patients because the HPC death triggers inflammation and mitochondrial damage, increasing reactive oxygen species further accelerating this process.17 This explains the progressive BMF.18 Functional reversion, where the pathogenic mutation is ‘reversed’ by a new somatic mutation leading to normal phenotypic function, occurs in ˜10–25% of patients. This gives rise to a reverted HPC, which has competitive advantage over the non-reverted FA HPC, allowing for spontaneous remission of the cytopenias.19 Mosaicism of the HPC in blood and marrow may result in false negatives on blood chromosomal fragility testing; therefore, in patients where there is high clinical suspicion, testing should be performed on skin fibroblasts. These patients remain at risk of MDS/AML by non-reverted HPC, and, since non-haematological tissue is not repaired, their risk of solid tumours remains unchanged.
The cumulative incidence of MDS and AML before the age of 40 years is 30% and 10%, respectively. The risk of cancer in FA is >500-times that in the normal population, with AML being the most frequent malignancy, although a wide range of solid tumours may also be diagnosed.20 Although FA patients are young when they present with AML, they do not typically have the favourable, reciprocal translocations associated with young patients with de novo AML, but instead they have multiple chromosome deletions and additions more typical of MDS/AML seen in older individuals or those with therapy-related disease. These include unbalanced gains in the long arms of chromosomes 1 and 3 and deletions in chromosomes 7, 11, and 21.21 Additionally, RUNX1 somatic mutations are seen in 20% of patients with FA and AML and are also associated with poor survival.22 When evaluating MDS/AML in younger patients, a history of progressive cytopenias and/or high-risk or complex cytogenetics should make the clinician consider an underlying diagnosis of FA.
The only current curative treatment for the haematological manifestations of FA is HSCT. The first attempt at HSCT for FA resulted in extreme TRM, but by the early 1990s studies showed survival could be improved by lowering the dosage of radiation and alkylating agents. A decade later, in the setting of lower dose total body irradiation (TBI) and cyclophosphamide, matched sibling donor (MSD) transplants, overall survival (OS) of >85% were reached, but with dramatically increased risks of non-haematological cancers in survivors.12,23,24 Current approaches use fludarabine-based reduced intensity conditioning (RIC), which allow for engraftment without high dose alkylating agents and TBI. This has further attenuated the TRM, and FA patients without MDS/AML now have ˜90% 5-year survival with a MSD transplant.25,26 A MSD who does not share the pathogenic mutation of both alleles is preferred. All full siblings should be human leukocyte antigen-typed and undergo FA testing regardless of phenotypic evidence of disease. A recent study was undertaken of FA patients without AML who were transplanted with an unrelated donor or haploidentical donor, without radiation and using a T cell deplete graft. This study had a 3-year OS of 80% without significant graft versus host disease (GVHD). Non-relapse mortality was 18%, infection being the most common cause.27 FA, HSCT, and GVHD are all independent risk factors for mucocutaneous human papilloma virus (HPV)-related squamous cell carcinomas (SCC); thus, FA patients who have been transplanted remain at high risk and require ongoing surveillance.28,29 The rationale of this study was to mitigate this risk of SCC by giving radiation-free conditioning and lowering the incidence of GVHD by T cell depletion. The last decade has seen major advancements in our understanding of the pathogenesis and the implications of diagnosis on treatment, resulting in improved patient outcome. Newer approaches include gene therapy trials, where the pathogenic mutation is corrected by in vitro transduction of the patient’s HPC. Treatment of overt leukaemia remains challenging, because treatment intensity and pre-transplant cytoreduction needed to cure AML can result in unacceptable non-relapse mortality; current strategies include sequential timing and/or addition of cytarabine.21,30 The treatment of AML is further evolving to include immunotherapy and targeted agents with less cytotoxicity and has the potential to further improve patient outcomes within the next decade.
DYSKERATOSIS CONGENITA
Dyskeratosis congenita (DC) is a heterogeneous disorder with variable modes of inheritance: X-linked, autosomal dominant, and autosomal recessive.1,31 Families may demonstrate genetic anticipation, where early generations have less severe disease and clinical manifestations are more apparent at earlier ages in subsequent generations.32 Patients with classic DC present with the triad of skin/nail dystrophy, reticular pigmentation of the skin, and mucosal leukoplasia in early childhood.33 Other manifestations include microcephaly, developmental delay, teeth abnormalities, lacrimal duct stenosis, exercise intolerance, oesophageal stenosis, SCC of the head, neck, and/or anogenital region, liver disease, hair loss, premature greying, osteoporosis, combined immunodeficiency, ataxia, and pulmonary arteriovenous malformations.1,33,34 More than 90% of individuals with DC will develop cytopenias by the age of 40 years. The classic presentation defines only a fraction of patients who primarily have mutations in the X-linked gene DKC1. Others with DC have BMF without mucocutaneous findings, isolated pulmonary and/or hepatic fibrosis, or are phenotypically silent. As with other iBMFS, this variation makes it a diagnostic challenge. Because the mucocutaneous and nail findings can mimic chronic GVHD, an atypical presentation of DC may be considered in patients transplanted for AA with what is thought to be severe GVHD.35 Pathologic mutations in 13 genes have been identified, all of which interfere with telomere maintenance; however, approximately one-third of DC remains genetically unclassified.31 Mutations in TINF2, RTEL1 (autosomal recessive form), and in the catalytic domain of DKC1 are associated with the more severe phenotypes; whereas mutations in TERC and TERT are often diagnosed later with BMF/AA and/or fibrosis. The diagnosis may be established by clinical findings and confirmed by telomere length measurements in leukocyte subsets, such as by flow cytometry fluorescence in situ hybridisation (flow-FISH).31,36 Flow-FISH is 95% specific and nearly 100% sensitive in patients with a telomere length <1% of age-matched controls.37
DC also demonstrates a premature ageing phenotype with early attrition of progenitors. Maintaining adequate telomere length is important in dividing progenitors in all tissues. Therefore, it is not surprising that most of the tissues severely affected have more actively replenishing progenitors, such as blood, skin, and mucus membranes. Pathogenic mutations in DC decrease the function of telomerase complex, disabling the cell from being able to maintain its telomere length and results in prematurely shortened telomeres.38 Telomeres are structures made from tandem DNA repeats and associated proteins that cap the ends of chromosomes and protect them from degradation. Telomere ends that have shortened below a critical length signal that a cell has reached the end of its replicative potential and the cell becomes senescent or apoptotic. In DC, progenitors lose full replicative potential, undergoing early attrition.
Patients with DC have malignant predisposition, particularly for SCC and MDS/AML. Compared to the general population, patients with DC have a 195-fold increased risk of AML, a 2,362-fold increased risk of MDS, particularly hypocellular MDS, and a 1,154-fold increase in the risk of head and neck squamous cell cancers.39 By the age of 50 years old, the risk of malignancy is ˜20–30%, for the majority solid cancers. As previously stated, cells with shortened telomeres activate p53 and undergo replicative senescence, but if the cell has developed a premalignant somatic mutation (such as in p53) then the cell will be unable to respond to these signals to arrest, and will divide until very short telomeres cause a telomere crisis. This has two different outcomes: an appropriate response of cell death, or the cell acquires a second hit, another somatic mutation which may cause immortalisation and cancer. Furthermore, shortened telomeres increase genomic instability due to end-to-end fusions and that genes involved in telomere maintenance are also involved in DNA repair.40 While DC is more rarely associated with MDS or AML in children compared to FA, it must be considered along with other iBMFS during the evaluation of young people with MDS/AML, particularly as serious treatment-related toxicity is often seen with standard cytotoxic leukaemia regimens.
The only curative treatment for haematological manifestations is HSCT. MSD transplant with conventional radiation-based conditioning is associated with high TRM.41 The clinical sequelae of some chemotherapy agents, radiation, and GVHD overlaps with the natural clinical progression of DC and are independent risk factors for SCC, restrictive lung disease, and liver disease. Treatment therefore amplifies the risk of secondary cancers. Patients with DC and pre-existing pulmonary fibrosis and/or hepatic cirrhosis are more susceptible to TRM. RIC-HSCT is preferred and the agents that cause lung and liver toxicity, such as busulfan and high dose TBI, should be avoided.42-45 The 5-year OS with this approach is ˜70%, with patients >20 years old having significantly worse outcomes than younger patients.43 If radiation is required, then lungs should be blocked from the field. The preferred donor in DC is a MSD and, because clinical findings may be absent, it is important to screen all potential family donors for the gene mutation and/or by telomere assay. The chance that a sibling is both fully matched and unaffected is ˜12%.
RIBOSOMOPATHIES: SHWACHMAN–DIAMOND SYNDROME AND DIAMOND–BLACKFAN ANAEMIA
Ribosomes are the complexes responsible for the translation of mRNA and are a critical component of cellular machinery. Gene mutations that result in ribosome dysfunction occur in several iBMFS.46 Like other iBMFS, there is clinical heterogeneity, but common findings include cytopenias, congenital anomalies, stress erythropoiesis, and a predisposition to cancer including MDS/AML. These syndromes are more easily diagnosed in a patient with a classic constellation of symptoms in childhood, but, because not all individuals will have the classic manifestations, some may escape diagnosis until later when they present with MDS.
Two of the classic syndromes are Shwachman–Diamond syndrome (SDS) and Diamond–Blackfan anaemia (DBA). SDS is a recessive disease due to biallelic mutations of SBDS. Affected children classically present in early childhood with exocrine pancreatic insufficiency, neutropenia, congenital anomalies, dystrophy, and skeletal abnormalities.47 Although neutropenia is the most common manifestation of SDS, all lineages can be affected. DBA is caused by heterozygous mutations of 12 ribosomal genes with variable inheritance patterns, including autosomal dominant with haploinsufficiency of RPS19 (most common), X-linked, and autosomal recessive. Patients with DBA present with pure red cell aplasia and congenital craniofacial, skeletal, renal, and/or cardiac anomalies, in the first year of life.48 Anaemia, reticulocytopenia, macrocytosis, and a marrow deplete of erythroid progenitors are diagnostic of DBA.1,48 Although there is no clear genotype-phenotype correlation for the severity of the cytopenia, the non-haematological manifestations tend to segregate with the pathogenic mutations. Thumb abnormalities are virtually absent in patients with RPS19 mutations but are frequently present in those with RPL5 or RPL11 mutations.1,49 The X-linked form is rare and only two families have been identified, but is unique in that the gene mutated is not a ribosomal gene but GATA1, encoding a transcription factor. These patients only express haematological manifestations, although not elevated ADA. Elevated ADA distinguishes patients with DBA from transient erythroblastopenia of childhood, so knowledge of this exception is important as the patients with GATA1 mutations are at risk of MDS/AML.50 Increasingly, these syndromes are being recognised in AYA who have had mild manifestations or spontaneous remission of the anaemia earlier in life and not recognised until they present with MDS/AML or another cancer. The cumulative incidence of MDS/AML in SDS is estimated to be ˜10% by the age of 20 years, most frequently involving deletions of chromosome 7. This is in contrast to the estimated occurrence of MDS/AML, thought to be <2% of patients with DBA, although this may be higher in those with GATA1 mutations.51,52 DBA patients also have an increased risk of other cancers with an overall risk of malignancy estimated to be 20%.52 HSCT is curative for patients with SDS and DBA who have severe haematological manifestations. Patients with SDS experience increased toxicity with transplant, and RIC-HSCT should be considered.53 Patients with DBA can have excellent outcomes with MSD-HSCT, and results with unrelated donor-HSCT are improving. Iron overload is a risk in patients that are heavily transfused from an early age and should be evaluated and treated prior to transplant. All potential related donors should be genetically screened.
GERMLINE MUTATION OF MASTER PROGENITOR REGULATORS: GATA1/GATA2/RUNX1
Like GATA1, GATA2 also encodes a transcription factor and germline mutations have been associated with cytopenias and predisposition to MDS/AML.54 GATA1 germline mutations cause a spectrum of cytopenias from isolated anaemia (DBA), to thrombocytopenia, or rarely pancytopenia. Germline mutations of GATA2 have historically been described as two classic immunologic syndromes. Emberger syndrome is described as lymphoedema, sensorineural hearing loss, warts (HPV-associated), low CD4/CD8 ratio, and MDS; monocytopenia and mycobacterial infection syndrome is described as monocytopenia and predisposition to viral/nontuberculous mycobacterial infections. Autosomal dominant inheritance or de novo mutations of germline GATA2 have a variable phenotype and other features may include neutropenia, lymphopenia (B lymphocytes and natural killer cells), BMF, MDS/AML, Epstein–Barr virus-associated cancers, pulmonary alveolar proteinosis, neurocognitive delay, and erythema nodosum.54-57 An elevated peripheral CD34 count is often present at the onset, but declines with declining blood counts. GATA2 is now recognised as the most common germline mutation to cause MDS/AML in younger patients. It has recently been reported to account for 9% of all primary MDS cases in children <18 years of age, and 37% of young patients (72% of patients ages 12–19) with monosomy 7 MDS.5 Therefore it is recommended that all young patients with primary MDS with monosomy 7 should undergo germline GATA2 testing. HSCT is the only curative option. Not only is AA, MDS, and/or AML an indication for transplant, transplant in patients with germline GATA2 mutations has also been shown to improve the underlying immunodeficiency and pulmonary alveolar proteinosis.
RUNX1 is a haematopoietic transcription factor that is upregulated after the progenitor cell undergoes megakaryocytic differentiation. Germline mutations of RUNX1 cause autosomal dominant familial platelet disorder with associated myeloid malignancies (FPD/AML).58 These patients often experience lifetime bleeding tendencies secondary to moderate thrombocytopenia and functional platelet defects. An estimated 35–40% of these patients will develop AML typically as an AYA; the median age of diagnosis is 33 years of age, although patients who develop AML in the first year of life through to their mid-60s have also been described. T cell acute lymphoblastic leukaemia has also been described in these families. Patients with dominant negative mutations have a higher propensity to develop leukaemia. Not all patients will present with bleeding and may go undiagnosed until the development of MDS/AML. This syndrome also appears to be associated with genetic anticipation.58-61
CONCLUSION
Phenotypic and genotypic variation of the iBMFS make diagnosis challenging. It is important for the clinician to maintain a high level of suspicion when evaluating younger patients with AA, MDS/AML, since treating with chemotherapy or HSCT without knowledge of an underlying diagnosis leads to poor outcomes. Table 1 outlines some of the findings that can suggest diagnosis of iBMFS. Taking a detailed family and personal history, careful physical exam, and thorough review of laboratory and cytogenetic findings can assist the clinician in determining when to pursue testing and genetic counselling referral.3 This will lead to better patient outcomes and allows adherence to syndrome specific guidelines.6-8 HSCT remains the mainstay of treatment for these patients and diagnosis is an important factor in choosing pre-transplant therapy, conditioning regimen (agents and intensity), donor type, GVHD prevention strategy, and survivorship screening.
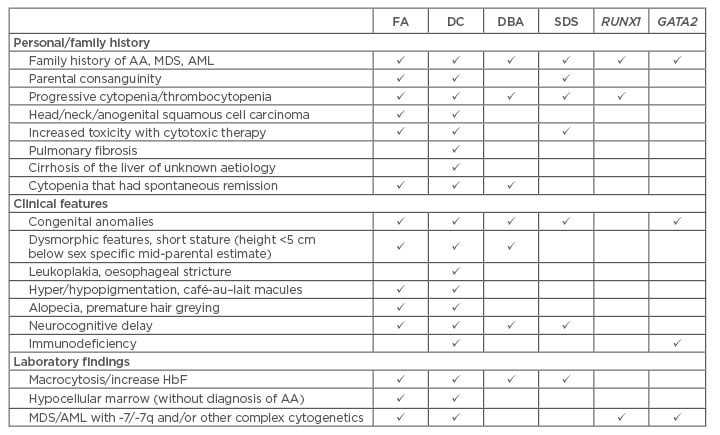
Table 1: When to consider evaluation for inherited bone marrow failure syndromes in a young patient with cytopenias, myelodysplastic syndrome, or acute myeloid leukaemia.
AA: aplastic anaemia; AML: acute myeloid leukaemia; DC: dyskeratosis congenital; DBA:
Diamond–Blackfan anaemia; FA: Fanconi anaemia; HbF: haemoglobin F; MDS: myelodysplastic syndrome; SDS: Shwachman–Diamond syndrome.





