
Abstract
Melanoma is the most aggressive and deadly form of skin cancer. The high rate of patient death is related to advanced melanoma metastasis, which usually occurs several months to years after the primary melanoma diagnosis. At an early stage, the melanoma tumour can be removed, therefore promoting a survival rate up to 99%. In this manuscript, we elucidate the tumour microenvironment factor, which is crucial for melanoma growth, proliferation, and metastasis. Melanoma is more resistant to traditional therapies, such as chemotherapy and radiotherapy; indeed, tumour-associated macrophages are often related to the worst prognosis. A better understanding of the melanoma microenvironment, including melanoma-associated fibroblasts and hypoxia-inducible factors, will enable researchers to develop drug-delivery systems with higher anticancer activity than current melanoma therapies available on the market. This review also covers macrophage targeting melanoma, such as macrophage colony-stimulating factor receptor inhibitors, C-C chemokine ligand 2 inhibitors, and vaccines combining αFAP-PE38 and melanoma associated antigens via lentiviral vectors. We also report a study using statins, which demonstrated long circulating liposome-encapsulate simvastatin reduced tumour-associated macrophage-mediated oxidative stress and production of the hypoxia-inducible factor 1α in tumours. In melanoma, xenografts may be treated with antiangiogenic agents targeting different angiogenic pathways, such as properdistatin, which selectively removes small diameter vessels and reduces the blood supply time. Sunitinib also plays a role in reducing the density of small and large diameter vessels, although it does not change the blood supply time. Considering all these factors holistically suggests that a better understanding of the melanoma microenvironment is crucial for the development of a novel and effective therapeutic approach.
INTRODUCTION
Melanoma is one of the most common types of cancer; in the USA, it is the 7th leading cancer in
women and the 5th in men. Over the last few years, the incidence of melanoma has increased
substantially, especially in women. This type of cancer is the most aggressive skin cancer, with a high rate of patient death due to advanced melanoma metastasis, which usually occurs several months to years after the primary melanoma diagnosis. This is despite a survival rate of up to 99% if a melanoma tumour is removed at an early stage.1 Fortunately, this paradigm has changed due to advances in new drug pathways and targeting in drug delivery systems, which may have a major impact on the development of immunotherapy and target therapy for melanoma cancer. The drugs most commonly used for the treatment of the metastatic effects of melanoma are ipilimumab and vemurafenib, both approved by the U.S. Food and Drug Administration (FDA), although both therapies have their limitations. These drugs play different roles in melanoma cells; ipilimumab can achieve durable benefits to the target cells by blocking the immune suppression of T cells that is induced by cytotoxic T lymphocyte antigen 4; however, 80–85% of patients do not respond to this therapy. On the other hand, vemurafenib can achieve rapid tumour regression by targeting melanoma cells harbouring BRAF Val600Glu mutations. A negative side of this drug is that patients may develop drug resistance after 6 months of treatment. This manuscript presents the melanoma network environment affecting melanoma development, focussing on tissue hypoxia, macrophages, and stromal fibroblasts.2 It is only possible to develop new drugs and strategies for melanoma therapy, prognosis, and diagnosis through a better understanding of how the tumour microenvironment can directly affect melanoma cancer progression.
SKIN AND MELANOMA MICROENVIRONMENT
The skin represents the largest organ of our body. Normal skin consists of two distinct layers: the epidermis and the dermis. The epidermis is the upper layer formed by keratinocytes, melanocytes, and Langerhans cells. Keratinocytes are the most abundant cell type in the epidermis layer and are responsible for producing the major structural protein of the skin using growth factors and keratin to maintain normal skin homeostasis. The keratinocytes also regulate and promote the proliferation of melanocytes through connexins, desmoglein-1, and E-cadherin.3 The epidermal-melanocyte unit is normally formed by one melanocyte surrounded by 5–8 keratinocytes. Melanocytes are responsible for producing pigments that contain melanin, called melanosomes. The major risk for melanoma development is the incidence and absorbance of ultraviolet (UV) radiation; upon UV exposure, the melanosomes are transferred from melanocytes to the keratinocytes. This mechanism can avoid DNA damage caused by UV radiation.4 In the skin there are antigen cells, namely, Langerhans cells, which are dendritic immune cells. The dermis is mostly formed by fibroblast cells, responsible for collagen production and release, and also for producing elastin; thus, maintaining the tight connection of the dermis and the epidermis through a basement membrane. In addition to fibroblast cells, there are also pericytes, adipocytes, macrophages, and vascular endothelial cells.
The melanoma microenvironment conditions differ from the normal skin and alter as the melanoma phases progress. A radial growth phase, followed by a vertical growth phase, occur in the early stages of melanoma. The last phase, which worsens the patient’s condition, occurs at the metastatic stage. When talking about the melanoma microenvironment it must be mentioned that it is highly heterogeneous and contains a variety of extracellular matrix (ECM) cells and noncancerous cells, such as fibroblasts, keratinocytes, and inflammatory cells. In this microenvironment, it is also possible to find growth factors produced by stromal cells and cancer cells. The interaction between the cancer cells and surrounding cells is very complex. The cytokines and growth factors produced by melanoma cells are responsible for recruiting many types of stromal cells, activating the tumour microenvironment, which can occur either directly or indirectly. When the growth is indirect, it affects many functions of other types of stromal cells, which play an important factor in melanoma initiation, progression, and, finally, metastasis. When the tumour is activated directly, the melanoma growth is promoted by activated stromal cells within the tumour microenvironment.5 In Figure 1 we can observe B16 murine melanoma cells.
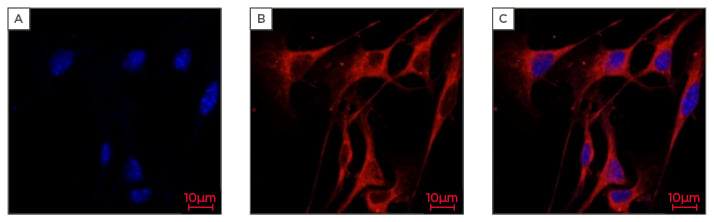
Figure 1: Confocal fluorescence images of B16 (murine melanoma cells).
The confocal images were acquired by Zeiss LSM800 Airyscan microscope (Zeiss, Oberkochen, Germany)using X63 oil immersion objective lens.
A) Stained with Hoechst to visualise the nuclei (blue); B) Cells stained with Phalloidin Rhodamine Actin to visualise cellular morphology (red); C) Multiple layers indicating both the nuclei (blue) and cellular morphology (red).
Deprived of adequate oxygen supply, hypoxia of the dermal environment takes place, promoting melanomagenesis.6 While the tumour grows, it requires nutrients, oxygen, and blood; as a consequence, an uneven vasculature distribution starts to form.5 Hypoxic tumour cells are often associated with worse prognosis, because this kind of tumour is more resistant to radiation and chemotherapies due to its peculiar biological properties and metabolic rates when compared to well-oxygenated tumours.7 Tumour aggressiveness and hypoxia are linked, as shown in recent studies that demonstrated the treatment of tumours in mice with antiangiogenic agents can lead to tumour hypoxia, therefore enhancing the invasion and migration properties.8,9 Tumour hypoxia and the tumour-stromal cells microenvironment can play an important role in melanoma skin cancer initiation, progression, and treatment resistance. Hypoxia-inducible factors (HIF) are present in solid tumours when the malignant tumour has a diameter >1 mm. Consequently, pockets of hypoxic regions can be visualised by immunohistochemistry with carbonic anhydrase,9,10 primonidazole,11 or antibodies. Normal oxygen level in the skin ranges from 1.5–5.0%. This concentration of oxygen is sufficient for the stabilisation of HIF-1α.12 At the dermal-epidermal junction, the melanocytes are physiologically hypoxic under normal conditions; therefore, hypoxia may play an important role in melanocyte transformation. In normal skin, the nuclear HIF-1α is detected, which suggests that HIF-1α is activated in melanocytes; in other words, HIF-1α is a microphthalmia-associated transcription factor (MITF) transcription target in melanocytes.13 MITF plays a crucial role in melanoma cancer progression, as it is a transcription factor that is involved in the regulation of genes that are related to migration, invasiveness, proliferation, survival, and metastasis of melanoma cells.
Known to induce tumour resistance to both radiation and chemotherapies, hypoxia has broad effects on tumour fate. Hypoxia encourages growth of cells with deficient functions of tumour suppressor p53,14 promotes genetic instability, reduces drug-induced apoptosis,15 and favours the growth of hypoxia-tolerant tumour cell clones.16 HIF plays an important role in melanoma tumour progression, chemotherapy resistance, and metastasis. Hypoxia can influence the metastatic progression, promoting a phenotype switch in melanoma cells from proliferative to invasive.17 Proliferative melanoma cells, once exposed to the hypoxic microenvironment, increase the invasive potential in an HIF-1α-dependent manner and downregulate melanocytic marker expression. Melanocytes are prone to oncogenic transformation under hypoxic microenvironment due to HIF-1α stabilisation. In vivo, under hypoxic conditions, HIF-1α-deficient melanocytes show an ability to delay and diminish transformation capacity in tumour growth. Very aggressive melanomas can occur due to the expression of nondegradable forms of the HIF-1α protein. The permissive environment of HIF-1α may act as a tumour promoter in cells by enabling HIF-1α stabilisation at low oxygen microenvironments in the skin, where tumour cell promoters have acquired oncogenic mutations and are genetically unstable.
MELANOCYTES AND MELANOMA MICROENVIRONMENT
The microenvironment of a growing tumour is enriched in exosomes that are secreted by cancer cells. Exosomes are organelle-like, lipid-bound membrane structures shed from cell membranes into body fluids and interstitial spaces during tumour development.18 Cancer cells can manipulate their microenvironment to optimise and achieve better growth conditions and metastasis in many ways.19 Melanoma skin cancer is a melanocyte-originated malignant disease with high propensity to metastasise. The course of metastasis involves multiple steps, including epithelial-to-mesenchymal transition (EMT); however, the term ‘EMT’ may not formally be attributed to melanoma. Although melanocytes do not belong to the epithelial lineage, primary melanocytes do express E-cadherin, which is required for their contact with keratinocytes in the basal layer of the epidermis. Different to many epithelial tissues, normal melanocytes express EMT-inducing transcription factor; this is considered a predisposing factor for melanoma with a high metastatic propensity.20 A novel therapeutic modality to target melanoma progression may be developed by an understanding of how tumour-derived exosomes contribute to the manipulation of the tumour microenvironment.21
HUMAN DERMAL FIBROBLASTS AND MELANOMA ASSOCIATION
During the development of a melanoma tumour, the fibroblast cells are activated by other types of stromal or tumour cells, playing an important role for tumour growth and progression. Cancer-associated fibroblasts are highly heterogeneous in terms of their functions and markers. The fibroblast-specific protein 1, commonly used as a marker for other cancers, is expressed in melanoma cells,22 and therefore it is possible that it may also be a good melanoma-associated fibroblast (MAF) marker. However, it cannot be detected in normal skin fibroblasts.23 Figure 2 shows human dermal fibroblast after a 24, 48, and 72-hour incubation period.
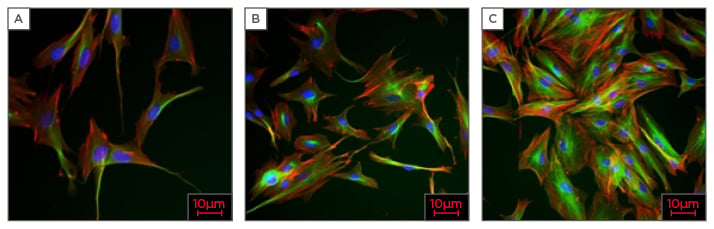
Figure 2: High content analysis of human dermal fibroblasts, magnification X20.
A) 24 hours after incubation; B) 48 hours after incubation; C) 72 hours after incubation.
Particularly in the Caucasian population, skin cancer is by far the most common oncological malignancy. Cutaneous malignant melanoma is one of the three most common types of skin cancer; the others are non-melanocytic skin cancers, such as squamous cell carcinomas, and basal cell carcinomas. The vascularisation of the tumour occurs naturally and the cells obtain all the required nutrients to grow until reaching the size range of 2–3 mm; this is called passive diffusion.24
Melanoma cell growth and survival rates can be highly affected by MAF. It was shown in xenograft models that co-injection of fibroblasts cells with melanoma cells increases tumour growth.25 In another study,26 it was also observed that, when coculturing normal fibroblast cells in association with melanoma cells, tumour growth was promoted. This growth was reported in the early-stage of the melanoma cells, but it has very little effect on melanoma metastatic cells.26 Some fibroblast cells can promote growth factors by overexpression of insulin, such as hepatic growth factor, basic fibroblast growth factor,27 and insulin growth factor-1, leading to growth of biological early stages and inducing the survival rate of melanoma cells by activation of beta-catenin and mitogen-activated protein kinase (MAPK).28 In addition, this growth factor also promotes both vascular endothelial growth factor and epithelial growth factor, both potent mitogens for the growth, proliferation, and microenvironment of melanoma cells. MAF also promote melanoma growth through glycosaminoglycan hyaluronan, also called hyaluronate, hyaluronic acid, or HA. The synthesis of HA (HAS1 and HAS2) in MAF can activate and enhance melanoma production of platelet-derived growth factor receptor (PDGF)-CC and PDGF-AA in dermal fibroblasts and melanoma cell-derived factor stimulates hyaluronan synthesis by regulating HAS2 through p38 and PDGFR-PI3K-AKT signalling.29 HA is one of the major components of the ECM and has been proven to promote melanoma tumour growth, angiogenesis, and metastasis.30
ADAM-9, a member of a family of proteases with a disintegrin and metalloprotease domain, is expressed at the tumour-stromal border. Studies in vivo and in vitro have found that stromal fibroblast-specific expression of ADAM-9 is responsible for melanoma proliferation and apoptosis, but it is controversial as to whether it exerts pro-tumour growth effects or tumour growth inhibitory effects.31,32
p53 is a suppressor of several types of tumours33 and can modulate tumour growth in stromal fibroblasts in a stromal cell-derived factor 1 dependent manner.34 Dysfunction of p53 occurs in >90% of melanoma cases.35 In melanoma, MAF play an important role in metastasis, as the invasive potential of human melanoma cells may alter the fibroblast gene expression.36
Metastatic effects are caused by the spread of primary cancer cells to distant sites of the body and are attributed to the majority of cancer deaths. In melanoma metastasis, it is possible to observe sequential steps:37,38 a) tumour cells are separated from the basement membrane, due to acquisition of an invasive phenotype, b) tumour cells can reach either a regional site or distant organs through haematogenous or lymphatic dissemination, c) tumour cells survive in a new metastatic organ such as bone, the liver, the brain, the lungs, etc.
Fibroblasts cells play an important role in melanoma angiogenesis, which consists of the formation of new blood vessels to supply nutrients for tumour growth, which is one step away from tumour cell metastasis. In a three-dimensional gel embedded with fibroblast type I collagen model and melanoma cells, normal human fibroblasts enable melanoma cells to induce angiogenesis.39 As a consequence of melanoma metastasis, MAF can enhance melanoma invasion and angiogenesis.
TUMOUR-ASSOCIATED MACROPHAGES
The microenvironment of melanoma tumours contains many types of inflammatory cells, including mast cells, neutrophils, dendritic cells, B and T cells, and macrophages. Tumour-associated macrophages (TAM) are the most abundant cell type among immune cells; they can interfere directly with the tumour microenvironment in numerous ways, such as remodelling the ECM, increasing tumour initiation and growth, promoting angiogenesis, and suppressing antitumour immunity through the production of growth factors, cytokines, reactive oxygen and nitrogen species, and chemokines.40-42 TAM are also linked with cancer resistance to therapies, such as immune therapy, target therapy, radiotherapy, and chemotherapy. The patient’s response to antitumour therapies can be predicated by the number of macrophages; the higher the number of infiltrating macrophages, the worse the prognosis in a variety of cancers, including melanoma.43 An inflammatory microenvironment plays an important role in every step of melanoma development and can be provided by TAM and highly inflamed tumours.
The association between melanoma and macrophages can be classified as:2
- M1 macrophages: Activated macrophages: the macrophage is polarised by proinflammatory factors, such as IFN-γ, and microbial agents, such as bacterial lipopolysaccharide. In this macrophage, reactive oxygen species are produced along with higher levels of nitric oxide, tumour necrosis factor α, interleukin (IL)-6, IL-12, and lower levels of the immunosuppressive cytokine IL-10. Many growth factors produced by M1 macrophages have a significant effect on promoting tumour growth and metastasis, such as IL-6 and tumour necrosis factor α.
- M2 macrophages: Alternatively activated macrophages: TAM resemble M2 macrophages and exert pro-tumour activity. M2 macrophages produce lower levels of IL-12 and a higher level of transforming growth factor (TGF)-β, IL-1 receptor antagonist (IL-1ra), IL-10, chemokine ligand (CCL)1, CCL18, and CCL22.
The WNT gene family consists of structurally related genes that encode secreted signalling proteins. These proteins have been implicated in oncogenesis and in several developmental processes, including the regulation of cell fate and patterning during embryogenesis. WNT Family Member 5A (WNT5A) is a protein-coding gene. Linnskog et al.44 published a study demonstrating a tumour-promoting role of increased WNT5A expression in malignant melanoma, sharing the ability to increase melanoma cell invasion. It reported pro-inflammatory cytokine IL-6 as a potential regulator of WNT5A expression. The inhibition of p38-MAPK completely blocked the IL-6-induced invasion of HTB63 cells. These findings suggest that recombinant WNT5A rescued the IL-6-induced invasion, indicating the inhibition of p38-MAPK activity and resulting in downstream inhibition of IL-6-induced cell invasion, related to impaired WNT5A expression.44
MELANOMA THERAPY BY MACROPHAGE TARGETING
Many approaches have been developed to target macrophages in melanoma, due to their multifunctionality and ability to promote tumour progression and metastasis. Studying the mechanisms of macrophages on tumour development has the potential to highlight possible options to treat the patient´s tumour.
Macrophage Colony-Stimulating Factor Receptor Inhibitor
Macrophage colony-stimulating factor (M-CSF) is the most potent growth factor and is highly expressed in melanoma cell lines. M-CSF is related to melanoma-associated macrophages, and activates multiple survival signals for macrophages, and binds to the M-CSF receptor (M-CSFR). Clinical trials of several M-CSFR inhibitors (M-CSFRi) for metastatic solid and advanced cancers, including melanoma, are underway. These trials are focussing on cancer therapy from the tumour microenvironment and cancer cells to tumour cells (Table 1). PLX3387, an M-CSFRi, was developed by Plexxikon Inc. (Berkeley, California, USA) and can increase the antitumour activity of BRAF inhibitors (BRAFi). Currently, Phase I of the trial is underway for patients diagnosed with melanoma. Anti-M-CSFR antibody, AMG 820, from Amgen (Uxbridge, UK) are under trial for advanced solid tumour therapy. The M-CSFRi, ARRAY-382, is also currently undergoing clinical trials for use in treating solid tumours.
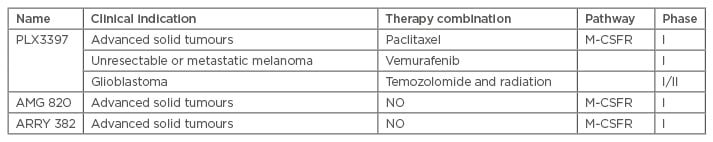
Table 1: Summary of drugs that target macrophages.
M-CSFR: macrophage colony-stimulating factor receptor.
Adapted from Wang et al.2
CCL2 Inhibitor
CCL2 plays an important role in macrophage survival and differentiation, and it is the most potent chemoattractant for macrophages. For melanoma therapy, some studies indicate that anticancer therapies in combination with CCL2 targeting are useful and vital.45 The expression of CCL2 can be decreased by the antitumour activity of BRAFi. De novo tumourigenesis and mouse melanoma xenograft models can achieve synergistic effects on tumour growth in combination therapy of an anti-CCL2 antibody and BRAFi.46
Liposome-Based Assay
Composed by bilayers of phospholipid, liposome is an artificially prepared vesicle structure. Drugs can be phagocytosed and recognised by macrophages when loaded in liposomes, providing efficient macrophage targeting. Liposomes have less toxicity than regular chemotherapy agents and can minimise the degradation of chemotherapy agents in serum.47 Liposome-encapsulated clodronate sulphate forces the macrophages to undergo apoptosis; the removal of macrophages leads to the angiogenesis inhibition and melanoma growth in a human melanoma xenograft model.48 Banciu et al.49 showed that liposomal prednisolone phosphate has an antitumour growth effect on deletion of TAM in melanoma models (mouse B16).
CURRENT STATUS OF TARGETING STRATEGY FOR MELANOMA-ASSOCIATED FIBROBLASTS, TUMOUR-ASSOCIATED MACROPHAGES, AND HYPOXIA-INDUCIBLE FACTORS
Recently, a therapeutically effective cancer vaccine has been developed for the treatment of melanoma, which needed to generate potent antitumour immune responses and at the same time overcome the tolerance mechanism mediated by the progression of the tumour itself. A novel immunotoxin was engineered, αFAP-PE38, which targets the fibroblast activation protein-expressing fibroblast within the tumour stroma. When researchers combined αFAP-PE38 and melanoma associated antigens via lentiviral vectors (termed LV-3Ag), it exhibited greatly enhanced antitumour effects on tumour growth. The mechanism of action was described as the potential combination to modulate the immune suppressive tumour microenvironment, activating the cytotoxicity of CD8+ T cells that are able to identify and destroy the malignant cells. Combining cancer vaccines with immunotoxin could be an alternative approach for patients with advanced cancer.44
Statins, used for lowering cholesterol, when used in higher doses (100–500-fold higher), possess antitumour activity with regard to B16F10 murine melanoma cells. Statins can have anticancer activity due to their pleiotropic action on key regulatory molecules (small GTP-binding proteins, such as Ras, Rac, and Rho) of intracellular signalling pathways responsible for cell inflammation, proliferation, angiogenesis, oxidative stress, and apoptosis.50 Long-circulating liposome-encapsulated simvastatin has an 85% higher anticancer activity than free simvastatin; it was reported that the antitumour activity of long-circulating liposome-encapsulated simvastatin is related to the presence of functional TAM in tumour tissue. The effect is based on the reduction of TAM-mediated oxidative stress and production of HIF-1α in tumours.51
Tumour hypoxia in R-18 human melanoma xenografts was reported to be treated with an antiangiogenic agent targeting different angiogenic pathways. Sunitinib may reduce the density of small and large diameter vessels whilst not changing the blood supply time, whereas properdistain selectively removes small diameter vessels and reduced the blood supply time. Both drugs were reported to inhibit angiogenesis; however, the drugs have had different effects on vascular morphology and function, and presented an extent of hypoxia in R-18 human melanoma xenografts.52
CONCLUSION
In this manuscript, we elucidated the cancer genetics to promote the development of new strategies for treatment of melanoma skin cancer, from the conventional therapies to target therapies. A better understanding of the melanoma tumour microenvironment can lead to a more effective therapy. Nowadays, several therapies targeting tumour microenvironments are undergoing clinical trials. The success of these trials may alter the approach of cancer therapy from tumour suppressor genes or specific protein-coding oncogenes, to targeted, less invasive, and much more effective therapies.






