
Abstract
Chemotherapy-induced mucositis is a common, often severe, side effect experienced by cancer patients during their treatment, which is a major challenge for successful anticancer therapy. As chemotherapy regimens evolve to include more efficacious agents, mucositis is increasingly becoming a major cause of dose-limiting toxicity and merits further investigation. This condition is characterised by both inflammation and loss of cells from the epithelial barrier of the gastrointestinal tract. This article reviews the current understanding of the inflammatory mechanisms behind chemotherapy’s toxic effect on the gastrointestinal tract and provides evidence that inflammation is a key factor behind gastrointestinal toxicity of chemotherapy. The authors discuss potential therapeutic targets that can contribute to mucositis treatment and prevention.
MUCOSITIS AS A MAJOR CHALLENGE FOR SUCCESSFUL ANTICANCER THERAPY
Mucositis is one of the most undesired side effects of antineoplastic chemotherapeutic and/or radiotherapeutic treatments.1 This condition is characterised by both inflammation and loss of cells from the epithelial barrier of the gastrointestinal (GI) tract. Clinically, mucositis is associated with various symptoms, such as severe GI pain, nausea, bleeding, severe diarrhoea, and starvation, which can lead to a reduction or interruption of antitumour treatment.2 All of these complications result in longer hospitalisation, increased cost to the healthcare system, and increased risk of mortality.
The current model for the development of mucositis suggests that there are five intertwined phases: initiation, upregulation and generation of messenger signals, signal amplification, ulceration, and healing (Figure 1).2 Briefly, in the initial phase, chemotherapy damages the DNA of epithelial cell progenitors and induces intense oxidative stress and cell death. In the second and third phases, there is an increase in cellular apoptosis and a progressive loss of cells from the crypt and the absorptive surface. An influx of inflammatory cells and increased production of inflammatory mediators in the mucosa are also hallmarks of these three phases. These events amplify the damaging process and lead to epithelial erosion. In the fourth stage, there is ulceration of the epithelium. This may result in the invasion of the submucosa layer by the indigenous microbiota and exacerbation of the inflammatory process. Changes in microbiota composition are common during mucositis development, resulting in dysbiosis,1,3 and they are important for the pathogenesis of mucositis. The last step is the recovery, in which cessation of the application of chemotherapy leads to restoration of the GI structure.4 Some of the molecular mechanisms involved in the progression of these pathologic phases have recently been described and will be detailed in the following sections of this manuscript, focussing on the contribution of inflammatory mediators to the progression of mucositis.
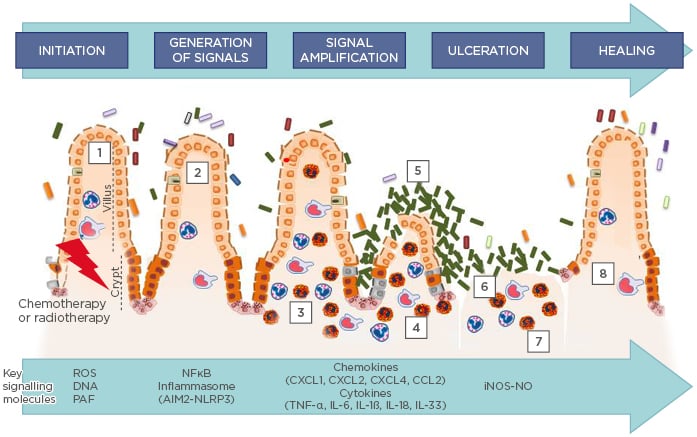
Figure 1: Current model of the pathophysiology of mucositis.
According to the current model, five phases are important in the pathophysiology of mucositis: 1) The generation of ROS, and DNA release from damaged cells and lipids mediators, like PAF during the initiation phase; 2) Activation of NFκB and inflammasome multiprotein assembly, followed by the induction of messenger molecules, such as TNF-α, IL-6, and CXCL1; 3) Treatment-related tissue inflammation and apoptosis during the upregulation/message generation phase, characterised by neutrophil, eosinophil, and macrophage influx to the lamina propria; 4 and 5) Microbiota-induced NFκB and inflammasome multi-protein assembly, via PAMP, which contributes to amplification of inflammation. The enhanced production of messenger molecules in the amplification and signalling phase, triggered by cytokine signalling, leukocyte influx, and dysbiotic microbiota, culminates in heightened inflammation and apoptosis (3 and 4), and discontinuity of the epithelial barrier resulting from apoptosis during the ulcerative phase (6), thereby promoting bacterial translocation (7). A spontaneous healing phase is initiated after chemotherapy cessation (8), characterised by intense cell proliferation.
iNOS-NO: inducible isoform nitric oxide synthase; NO: nitric oxide; PAF: platelet activating factor; PAMP: pathogen-associated molecular patterns; ROS: reactive oxygen species.
Adapted from Sonis.2
INFLAMMATORY MECHANISMS ASSOCIATED WITH THE AMPLIFICATION OF TISSUE INJURY DURING CHEMOTHERAPY-INDUCED MUCOSITIS
Mechanisms involved in mucositis pathogenesis are complex and are not limited to the direct epithelial damage induced by chemotherapy.5 Sonis et al.6 demonstrated that there is marked endothelial cell injury following exposure of the oral mucosa to antineoplastic therapy. This event precedes any detectable changes in the epithelium.6 Other studies have provided evidence that damage to GI stem cells is a consequence of extensive microvascular injury.7 Wherever the primary site of cellular injury, both culminate in the promotion of inflammation, a prominent component in mucosal damage during antineoplastic therapy, as highlighted in the model for mucositis development depicted above.1 Some of the molecular mechanisms involved in the progression of each of these steps are discussed below.
Reactive Oxygen Species and Inflammasomes
According to the model introduced by Sonis et al.,2 the primary inducer involved in unleashing mucosal injury upon chemotherapy is the production of reactive oxygen species (ROS), which is secondary to the chemotherapy-induced DNA damage. ROS are believed to cause direct cellular damage and tissue injury due to modifying several cellular structures.8 Hence, the authors have demonstrated that there is intense oxidative stress during irinotecan-induced mucositis in mice.9 This oxidative stress was shown to be dependent on NADPH-oxidase (NOX) activity, as Gp91phox-deficient mice and other animals models treated with a NOX inhibitor reverted irinotecan-induced oxidative stress. Importantly, NOX inhibition, by genetic or pharmacologic approaches, was able to prevent inflammation progression and mucositis development in the gut upon irinotecan treatment, implicating NOX2-derived ROS in chemotherapy-induced mucositis. In addition to NOX2-expressing leukocytes, it was demonstrated that enterocytes are a source of ROS production after chemotherapy and that the treatment of mice with the ROS scavenger fullerol leads to diminished inflammation and disease manifestation in irinotecan-treated mice.10 Other work has suggested that NOX1 plays an important role in the pathogenesis of 5-fluorouracil (5FU)-induced intestinal mucositis.11 In NOX1-deficient mice, the severity of mucositis was significantly reduced, particularly with respect to crypt disruption. This result was associated with attenuated production of ROS when compared to wild-type mice. NOX1-derived ROS production following administration of 5FU promotes the apoptotic response through upregulation of inflammatory cytokines.11 Radiation-induced mitochondrial dysfunction and ROS production were also shown to be involved in oral mucositis promotion in rats and preventing mitochondrial ROS production with melatonin was effective in preventing mucositis development.12 Altogether, these findings support the idea that chemotherapy-induced ROS production by several sources leads to tissue inflammation and mucositis induction.
The mechanisms involved in ROS-mediated amplification of the inflammatory response during chemotherapy-induced mucositis are still under investigation, but two pathways play major roles: the activation of NFκB and assembly of the multiprotein complexes called inflammasomes. Hence, chemotherapy and radiotherapy treatment are able to activate NFкB in epithelial cells, mesenchymal cells, endothelial cells, and macrophages, and its activation is believed to be secondary to chemotherapy-induced oxidative stress.1 NFкB plays a central role in triggering multiple proinflammatory signalling pathways, such as cytokine and chemokine synthesis.1,13,14 In this regard, Logan et al.13 demonstrated that expression and activation of NFкB precedes the peak of proinflammatory cytokine production in chemotherapy-induced mucositis. In another study, mice treated with a NFкB inhibitor showed diminished tissue damage, decreased IL-1β production, and neutrophil accumulation in the bowel after administration of 5FU.14 These findings reinforce the hypothesis that NFкB activation is central to the mucosal inflammation induced by chemotherapy and radiotherapy.
ROS production during chemotherapy also activates inflammatory mediator production through activation of the multiprotein complex inflammasome. Inflammasome assembly involves the oligomerisation of several proteins, including NOD-like receptors, such as NLRP3; adaptor proteins, such as ASC; and inflammatory caspases, such as caspase-1. The activation of this complex leads to induction of the caspase activity and the maturation of IL-1β and IL-18.15 In murine models, there is marked inflammasome activation in the gut, which is assessed by increased cleaved caspase-1 levels upon irinotecan-induced mucositis induction. NOX2-derived ROS were necessary for inflammasome activation in this system.9 Also, in radiation-induced oral mucositis, mitochondria-derived ROS are involved in NLRP3 inflammasome activation.12 Importantly, mice deficient for the ASC and caspase-1 inflammasome components were protected from mucositis development after irinotecan treatment,9 implicating this pathway in the amplification of inflammation and tissue injury during chemotherapy-induced mucositis. The cytokines IL-1β and IL-18 are well-known NFκB activators,16 implying that ROS-driven NFκB activation may be, in part, dependent on inflammasome assembly. Therefore, inflammasome activation seems to be pivotal for chemotherapy-induced mucositis.
Another implication for the inflammasome pathway in mucositis has been shown by Lian et al.,17 who highlighted the activation of AIM2 by genomic DNA released from the intestine. Double-strand DNA from germinative cells of the intestine is secreted in exosomes after irinotecan treatment; this DNA promotes IL-1β and IL-18 maturation in an AIM2-dependent manner, leading to mucositis and diarrhoea. Abrogation of AIM2 signalling, either in AIM2-deficient mice or by a pharmacological inhibitor, such as thalidomide, significantly reduces the incidence of drug-induced diarrhoea.17
Cytokines
It has been demonstrated that several proinflammatory cytokines and chemokines play prominent roles in the pathogenesis of mucositis. Several studies have shown elevated production of the inflammasome-dependent cytokines IL-1β and IL-18 during clinical and experimental chemotherapy-induced mucositis.9,16,18-21 In fact, the deficiency for IL-18 or treatment with IL-18BP or IL-1RA (which block IL-18 and IL-1 activity, respectively), resulted in a marked reduction in tissue inflammation and mucositis development.9,16-20 Furthermore, antibody neutralisation of IL-1 also prevented mucositis and epithelial tight junction dysfunction and alleviated mucositis in mice.16
In addition to promoting inflammation, IL-1β enhances mucositis development by triggering apoptosis of intestinal crypt epithelial cells via p53-mediated upregulation of p21 and p27.16 IL-1β apoptotic activity was blocked by IL-1RA administration.16 Altogether, these findings provide compelling evidence that IL-1β and IL-18 production are of pivotal importance for mucositis development.
IL-33, secretion of which is influenced by inflammasome activation, is produced by the intestinal epithelial cells of mice treated with irinotecan. Treatment of mice with an anti-IL-33 antibody or IL-33 receptor antagonist or deletion of the receptor for this cytokine resulted in attenuation of intestinal injury after chemotherapeutic intervention.21
In addition to inflammasome-derived cytokines, other inflammatory cytokines, such as TNF-α, IL-6, IL-4, and the chemokines CXCL1, CXCL2, CXCL4, and CCL2, are also associated with the development of intestinal mucositis induced by chemotherapy.18,21-26 Hence, inhibition of TNF-α and CXCL1 production with pentoxifylline or thalidomide resulted in a decrease in the pathogenesis of mucositis induced by irinotecan.18 Furthermore, knockout of IL-4 efficiently prevented the pathological alterations of 5FU-induced mucositis in the duodenum of the mice.18
CXCL1, CXCL2, and CCL2 are induced during mucositis in an IL-33-dependent pathway and promote neutrophil and macrophage recruitment, respectively, to the mucosa of the small intestine.21 CXCL4 and its receptor CXCR3 were confirmed, at both the genomic and protein level, to be regulated during 5FU-induced mucositis.24 CXCL4 neutralising monoclonal antibody decreased the incidence, severity, and duration of chemotherapy-induced diarrhoea and reduced apoptosis of the crypt epithelia by suppression of the 5FU-induced expression of p53 and BAX through its receptor, CXCR3. CXCL4 activates the phosphorylation of p38 MAPK, which mediates the stimulated expression of p53 and BAX, and results in the ultimate activation of caspase-8, 9, and 3.24 Therefore, CXCL4, through activation of CXCR3 receptor, fosters mucositis development by favouring epithelial cell apoptosis. Altogether, the above-detailed studies reinforce the concept that different types of proinflammatory cytokines might be important to the establishment of mucositis by maintaining and amplifying the inflammatory response.
In addition to cytokines, some studies have demonstrated the involvement of platelet activating factor (PAF), a leukocyte-mobilising lipid mediator in mucositis.27,28 Hence, patients with oral mucositis showed increased levels of PAF in the saliva.27 Mice-deficient for the PAF receptor were protected from 5FU-induced mucositis, with reduced production of TNF-α, CXCL1, and IL-1β,28 suggesting that this mediator might be involved in recruitment of the cells responsible for production of these proinflammatory mediators.
Leukocytes
Chemotherapy-induced cytokines and chemokines promote large infiltration of leucocytes into the lamina propria.1,9,20,29,30 More recent studies have confirmed that neutrophils,20 eosinophils (Arifa et al., manuscript in preparation), and macrophages21 play an important role in the pathogenesis of mucositis.
Several studies have indicated that neutrophil accumulation in the gut during mucositis contributes to tissue injury.9,18,21,23,25 Guabiraba et al.21 showed that neutrophils are highly activated in the blood 3 days after irinotecan treatment in mice. The concentration of CXCL1 was elevated in the serum at the same time. Furthermore, neutrophils accumulated in the small intestine were found to express the CXCR2 chemokine receptor. In addition, it was shown that inhibition of neutrophil influx into the intestine during treatment with irinotecan, by anti-Ly6G antibody or CXCR2 antagonism, resulted in attenuation of inflammation and intestine injury.
In addition, several studies have indicated that inhibition of cytokine action is associated with a reduced neutrophil accumulation in the gut during mucositis, thereby reducing tissue damage.9,18,20,21,23,25 Pentoxifylline, a methylxanthine derivative that reduces the expression of TNF-α, IL-1β, and IL-8, was associated with reduced myeloperoxidase activity and injury in the small intestine following chemotherapy- induced mucositis.18,23 Similar results were found by this group for thalidomide, an inhibitor of TNF-α production. IL-18, IL-33, or caspase- knockout mice or IL-18BP treatment clearly decreased neutrophil influx and injury in the intestine,20,21 as well as IL-1RA treatment in irinotecan-induced mucositis.9 Altogether, these results evidence that granulocyte recruitment to the mucosa during mucositis plays an essential role in tissue injury and disease development.
In addition to granulocytes, there is evidence to suggest that macrophages perform an important role in chemotherapy-induced mucositis.20 The number of F4/80-positive macrophages was markedly increased in irinotecan-treated mice. Moreover, these cells were identified as the major producers of IL-18 in the mucosa after irinotecan treatment. Taking these findings into consideration, it is reasonable to suggest that both granulocytes and macrophages are key agents for the pathology underlying chemotherapy-induced mucositis.
The nitric oxide (NO) synthases (NOS) plays a relevant role in the mucositis pathogenesis.23,31-33 This enzyme converts L-arginine and oxygen into L-citrulline and NO. NO is an important effector molecule produced by neutrophils and macrophages.34 Cytokines have been shown to stimulate the expression of the inducible NOS isoform 2 (NOS2) with consequent production of NO. This gas has antimicrobial activity and immune modulating and cytotoxic action toward the adjacent host tissues,35 resulting in pain, tissue lesions, and apoptosis.36-38 In radiotherapy and chemotherapy-induced mucositis, there are increases in NOS2 enzyme expression and activity in both the intestine and mouth mucosa.23,31,32,39 Additionally, mice treated with aminoguanidine, a selective NOS2 inhibitor, and NOS2-knockout mice have shown a reduction in radiotherapy and chemotherapy-induced mucositis due to reduced NO production.23,39 In summary, NO, released by neutrophils and macrophages, contributes to upregulation of inflammation and chemotherapy-induced injury.
INVOLVEMENT OF THE INTESTINAL MICROBIOTA IN MUCOSITIS
The authors and other research groups have demonstrated that host ability to mount an appropriate inflammatory response upon tissue injury is dependent on the intestinal bacterial colonisation.40-43 Germ-free (GF) mice are more resistant to radiotherapy and chemotherapy-induced mucositis;44-46 hence, GF mice are markedly resistant to lethal radiation and irinotecan-induced mucositis, and have shown a reduced number of apoptotic cells compared to conventional mice.44-46 GF mice treated with irinotecan presented with a reduced neutrophil and eosinophil accumulation and reduced levels of proinflammatory cytokines in the intestine, such as IL-1β and TNF-α, compared to irinotecan-treated conventional mice.46 These results suggest that the intestinal microbiota plays a key role in increasing chemotherapy and radiotherapy-induced intestinal toxicity, a phenotype that might be explained by the known modulatory role played by the microbiota over host inflammatory responsiveness41,43 or by directly exacerbating tissue injury induced by antineoplasic treatment due to bacterial colonisation of eroded intestinal tissue.
Based on the Sonis mucositis model, colonisation of the eroded mucosa by bacteria, fungi, and viruses occurs mainly at the ulceration phase and precedes secondary infections.2,6 Indeed, there is a higher abundance of microbiota in the ulcerated epithelium compared to the intact epithelium.47 These findings suggest that bacteria present on the ulcer surface are active contributors to the mucositis process.
However, only 8% of new patients with cancer will be at high risk of developing ulcerative mucositis. The majority of the new cancer patients are in the intermediate-risk category of developing mucositis, in which on average between 20% and 49% will develop ulcerative mucositis.6 Intestinal microbiota may be important contributors not only to the ulcerative phase of mucositis but also to the other stages of the disease.33,48 Furthermore, products of bacteria, such as metabolites and pathogen-associated molecular patterns (PAMP), may exert a chemotactic activity on leukocytes and stimulate them to further secrete proinflammatory cytokines, which does not depend on mucosa ulceration. PAMP are subject to innate immune monitoring by pattern recognition receptors. For example, cell wall components, such as lipopolysaccharide and β-glucan or flagellar components, are recognised as PAMP and trigger proinflammatory and antimicrobial responses in immune cells.49 The sensing of microbial products by these sentinel receptors could be involved in mucositis progression.
Indeed, some pattern recognition receptors, such as Toll like-receptor (TLR)2, 4, 5, and 9, and the receptors involved in inflammasome activation were reported to be involved in the pathogenesis of mucositis.9,17,33,50-52 TLR2, TLR9, and MyD88 signalling have also been implicated in doxorubicin and irinotecan-induced mucositis in mice; mice deficient for TLR2 and MyD88 expression were protected from mucositis development.33,53 TLR4 expression in the gut is induced upon administration of 5FU in the mouse model,54 but whether TLR4 activation is involved in mucositis progression has not been evaluated. Treatment with TLR5 agonists during radiotherapy decreased numbers of apoptotic cells in the GI mucosa, alleviated mucositis development, and improved survival of mice and primates.55,56 These studies strongly suggest that TLR5 activation by exogenous agonist administration is efficient at protecting the gut mucosa from damage. Altogether, these findings suggest that TLR and their activators, such as PAMP, play an important role in mucositis pathogenesis.
Changes in the microbiota, called dysbiosis, might promote dysregulation of the immune system.57-59 Dysbiosis also plays a relevant role in chemotherapy-associated mucositis.48,60 Several studies have demonstrated that chemotherapeutics exert a detrimental effect on the intestinal microbial composition. Shifts in microbiota composition differ along the GI tract and according to the chemotherapy regimen.3,9,61 Overall, dysbiosis during anticancer treatments leads to a reduction in the diversity and richness of the bacterial community.62 These alterations in microbiota composition usually coincide with the development of chemotherapy-induced mucositis in humans and in animal models.3 Altogether, these studies demonstrate that there are major changes in the microbiota during chemotherapy-induced mucositis and these shifts are potentially involved in the development of mucosal tissue injury.
There is evidence that the indigenous microbiota impact the development of chemotherapy-mediated mucositis. Some of the mechanisms involved in disease promotion by the gut microbial community are still under evaluation, but it is plausible to suggest that dysbiotic microbiota and their structural andmetabolic products foster mucositis development, at least in part, by amplifying the inflammatory mechanisms responsible for tissue injury progression.
POTENTIAL THERAPEUTIC TARGETS FOR MUCOSITIS TREATMENT
A better understanding of the pathogenesis of chemotherapy or radiation-induced mucositis is required to develop and implement optimal preventive and curative approaches for patients with this condition. Overall, the studies discussed so far provide extensive evidence that inflammation takes a central role in the pathogenesis of chemotherapy-induced mucositis. The knowledge gathered from these studies support the idea that interfering in inflammatory pathways may be efficient at alleviating mucositis development (Table 19,10,14,18,20,21,27,31) in a time-specific manner.
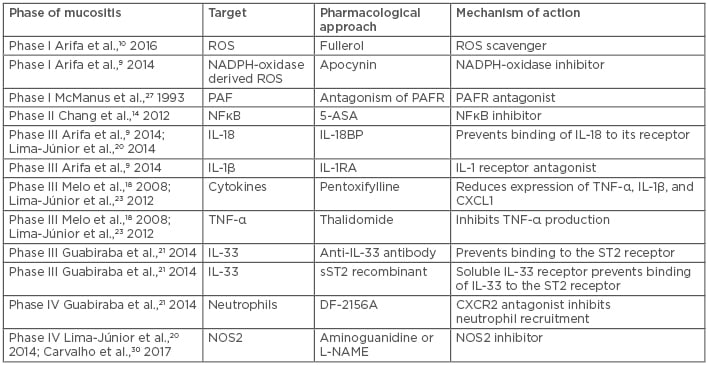
Table 1: Therapeutic targets that have been tested for the treatment of experimental intestinal mucositis in mice.
NOS: nitric oxide synthase; PAF: platelet activating factor; PAFR: platelet activating factor receptor; ROS: reactive oxygen species.
In this context, ROS, PAF, and NFκB inhibition are potential therapeutic targets during the initial phases of mucositis. As demonstrated in Figure 1, ROS and PAF, followed by the activation of NFκB, are the first signals released after the start of the chemotherapy or radiotherapy treatment. Several antioxidant agents have recently been investigated in the prevention of mucositis.63-65 A recent study has shown that the antioxidant N-acetyl cysteine was efficient at decreasing the frequency of severe oral mucositis in patients.63 Therapeutic targeting of these molecules may prevent the triggering of the entire response that leads to mucositis.
In addition, inhibition of the inflammasomes or the actions of their derived cytokines, IL-1β and IL-18, may prevent mucositis development in the second phase. Inhibiting cytokines and chemokines is a potential therapeutic strategy, especially at subsequent steps. Indeed, a nonsteroidal anti-inflammatory drug, benzydamine, that inhibits the production of proinflammatory cytokines such as IL-1β and TNF-α, is recommended for prevention of oral mucositis during anticancer treatment.66 Drugs targeting IL-1β and TNF-α are available, having been approved for use in other conditions, and could provide initial targets for alleviating inflammatory alterations during mucositis. Intervening in the recruitment of leukocytes and their effector molecules (NOS2 and NO) may also have a beneficial effect, but only in later stages of the disease and may have little effect on the maintenance of the initial events of the disease.
However, some anti-inflammatory compounds, such as pentoxifylline and misoprostol, were inefficient at preventing mucositis.66 These results clearly suggest that the medical community is far from comprehensively understanding this complex toxic side effect. A possible explanation for this discrepancy may be the dual effect played by inflammatory signalling during anticancer treatment: it is important for both the side effects and some of the tumouricidal properties of chemotherapy and radiotherapy, such as mucositis. Future research must also evaluate whether inflammatory molecules have an effect on the tumouricidal activity of the relevant chemotherapeutic drugs; a subject that will impact the translation of the findings discussed here to the clinic.
Finally, by influencing the expression and release of immune effector molecules, the maintenance of commensal intestinal microbiota is a potential target for homeostasis and health in the intestinal tract during anticancer therapy. Indeed, several studies have demonstrated the beneficial effects exerted by probiotics, symbiotics, and prebiotics in rodent models and in humans.66 In conclusion, the studies involving mucositis have highlighted several potential therapeutic targets. However, it is still necessary to advance these studies to allow new findings to be applied in clinical treatments.






