Abstract
There is a strong causal link between chronic diabetes, inflammation, and secondary osteoporosis; this link is also often discussed as the diabetic bone disease development process. In this process, where systemic chronic inflammation and insulin resistance (IR) are key contributing factors, diabetes directly disrupts bone remodelling, leading to bone weakness and an increased risk of fractures. Moreover, as IR and low-grade inflammation are also associated with both obesity and metabolic syndrome, diabetes and diabetic bone disease are attracting attention due to their increasing prevalence with age in both sexes, particularly in postmenopausal women. This brief review, highlighting the role of chronic systemic inflammation, IR, and metabolic interactions in the relationship between Type 2 diabetes and secondary osteoporosis, focuses on summarising current developments to provide references for clinical practice in light of recent research data and current literature. Furthermore, increased knowledge regarding diagnostic approaches can contribute to reducing the occurrence of frailty fractures in diabetes through early diagnosis and interventions.
Key points
1. The synergistic effects of insulin resistance and chronic systemic inflammation in diabetic bone disease necessitate a disease-specific management approach.2. Given the characteristics of diabetic bone disease, comprehensive risk assessment may help to prevent osteoporotic fractures.
3.Novel therapeutics and interventions may support targeted and personalised fracture prevention strategies.
INTRODUCTION
There is a complex pathological relationship between diabetes and osteoporosis, two of the most common chronic diseases worldwide. The interaction of disease-specific and systemic factors (such as premature ageing, chronic inflammation, and nutritional and metabolic disorders) is among the causal factors involved in skeletal remodelling, which varies from high bone turnover to abnormally low bone turnover. Chronic kidney disease, rheumatoid arthritis, Cushing’s disease, Crohn’s disease, and diabetes are among the leading chronic diseases that may contribute to this variability.1,2 Type 2 diabetes (T2D) in particular, which is characterised by hyperglycaemia, insulin resistance (IR), and chronic low-grade inflammation, is one of the major causes affecting bone health and leading to secondary osteoporosis.3 Remarkably, individuals with T2D face a 20–30% higher overall risk of fractures compared to the general population, with postmenopausal females facing an even more pronounced risk of short-term fractures. Studies have shown a stronger link between T2D and fracture risk, particularly lower extremity fractures, in women and those who have had T2D for a longer period.4
IR has a complex and often detrimental effect on bone mass at different stages of life, transitioning from a potential driver of poor bone development in youth to a contributor to increased fracture risk in older age. Although initially associated with higher bone mineral density (BMD) due to hyperinsulinaemia acting as an anabolic agent, chronic IR ultimately weakens bone quality and increases fracture risk by disrupting normal bone remodelling. It is associated with lower bone mineral content and reduced bone development during critical growth years, particularly in boys. It acts as a negative influence on peak bone mass attainment, increasing the risk of osteoporosis later in life. In the middle age, this relationship is typically ‘two-phased’. Low levels of IR may not adversely affect bone density, but rapidly increasing IR in young adulthood or during the menopausal transition is associated with faster and accelerating bone loss.5-7
Decreased bone metabolism, chronic inflammation, oxidative stress, and fat accumulation in the bone marrow emerge as key mechanistic factors in inflammation-related IR and diabetic bone disease.8 Chronic systemic inflammation, IR, and secondary osteoporosis are interrelated conditions that form a complex, self-perpetuating vicious cycle and frequently lead to serious musculoskeletal disorders. Notably, the increasing incidence of this triad in both metabolic and chronic inflammatory diseases, such as obesity, metabolic syndrome, and T2D, is a noteworthy phenomenon.9,10
THE CLINICAL IMPLICATIONS OF INFLAMMATION-DRIVEN IR IN BONE HEALTH
The interplay of inflammation and IR serves as a primary driver of diabetic bone disease or secondary osteoporosis, resulting in significant clinical implications, including impaired bone quality despite normal or high BMD, accelerated bone resorption, and high susceptibility to fragility fractures. The chronic, low-grade inflammation associated with IR (common in T2D) disrupts the normal bone remodelling cycle by promoting bone-resorbing osteoclasts while inhibiting bone-forming osteoblasts. The following subheadings and brief descriptions summarise the key pathophysiological examples leading to clinical outcomes and increased fracture risk in diabetic bone disease (Figure 1).1,6,8,11
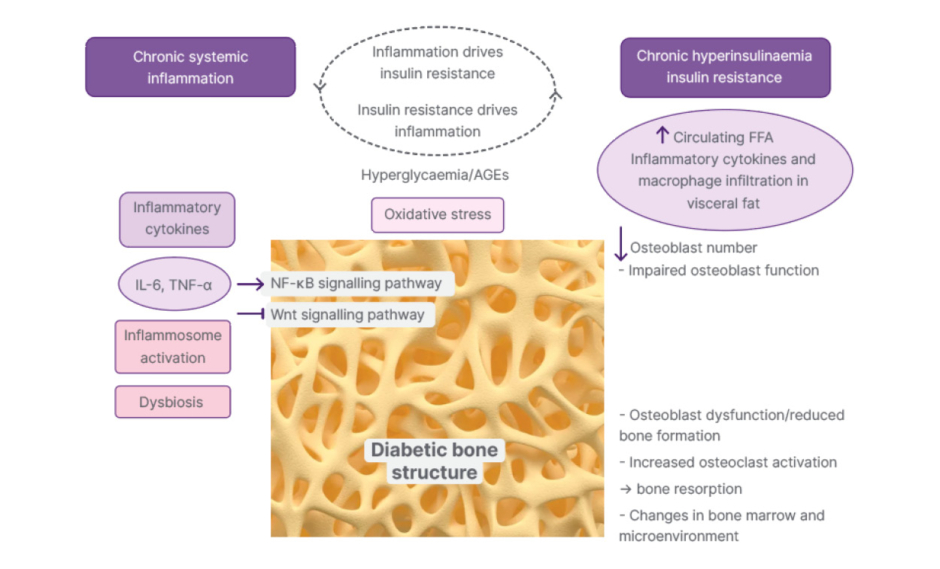
Figure 1: Pathogenesis of the synergy between chronic inflammation and IR.
Advanced glycation end products inhibited osteoblast differentiation. IL-6 and TNF-α can promote the proliferation and differentiation of osteoclast precursor cells into mature osteoclasts and accelerate the process of bone resorption.
AGE: advanced glycation end product; FFA: free fatty acids; IR: insulin resistance; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; Wnt: wingless-related integration site.
Paradoxical Fracture Risk
Patients with T2D experience a paradoxical increase in fracture risk, often referred to as the ‘diabetic bone paradox’, where bone fragility occurs despite having normal or elevated BMD. Whilst standard osteoporosis typically involves low bone mass, T2D-related bone disease is characterised by reduced bone quality, altered microarchitecture, and low bone turnover.
Impaired Bone Formation (Low Bone Remodelling)
IR prevents osteoblasts from responding to insulin, which is required for normal bone turnover, leading to decrease bone formation rates and impaired fracture healing.
Increased Bone Resorption (Osteoclast Activation)
In T2D, levels of pro-inflammatory cytokines (particularly TNF-α and IL-6) rise, and this directly stimulates the differentiation and activation of osteoclasts, thereby accelerating bone resorption.
Elevated Sclerostin Levels
Chronic inflammation increases the expression of sclerostin, a glycoprotein that inhibits bone formation by blocking the wingless-related integration site (Wnt) signalling pathway, which is essential for bone formation.
Advanced Glycation End Products Accumulation
Hyperglycaemia and inflammation promote the accumulation of advanced glycation end products in the bone matrix, making collagen brittle and less elastic, which increases fracture risk, especially in the cortical bone.
Bone Marrow Adiposity
Chronic inflammation and IR cause a shift in the differentiation of bone marrow stem cells, leading to the formation of more adipocytes and fewer osteoblasts. This, in turn, reduces bone density and quality.
Impaired Fracture Healing
Diabetic bone disease leads to an increased incidence of non-union fractures due to impaired osteoblast function and poor vascularisation, as well as prolonged healing times.
CLINICAL ASSESSMENT AND DIAGNOSTIC APPROACHES
In the diagnostic approach to diabetic bone disease, standard bone densitometry and imaging systems are prominent. Patients with chronic inflammation, T2D, and IR should be actively screened for bone health, as they are at high risk of undiagnosed secondary osteoporosis. To diagnose both diabetic bone disease and potential secondary osteoporosis, it is necessary to assess bone quality and integrity using imaging techniques and biomarkers. Dual-energy X-ray absorptiometry (DXA), the gold standard for BMD measurement, is useful for detecting osteoporosis, but it may not fully capture changes in bone quality associated with diabetes.12 Whereas, high-resolution CT provides detailed images of bone microarchitecture, including trabecular and cortical bone.13
The Trabecular Bone Score (TBS) is an advanced tissue analysis method that assesses bone microarchitecture using DXA data obtained from the lumbar spine.14 Unlike BMD measurements, which accurately measure bone mass, this method, which assesses bone quality, is preferred for evaluating and predicting the risk of fragility fractures independently of DXA.15 Whilst standard DXA only measures areal BMD, high-resolution peripheral quantitative CT (HR-pQCT) can evaluate both trabecular (inner) and cortical (outer) microarchitectural breakdown, which is often the primary cause of fragility fractures. HR-pQCT is a non-invasive, low-dose 3D imaging tool that allows clinicians to visualise and measure bone microstructure and volumetric density at peripheral sites like the radius and tibia. In the context of secondary osteoporosis, bone loss triggered by underlying conditions like chronic kidney disease, endocrine disorders, or medication use (e.g., glucocorticoids), HR-pQCT provides vital clinical insights that standard testing often misses.16,17
Studies have shown that high-resolution CT can identify trabecular bone loss and structural changes, which are critical in the assessment of diabetic bone disease.8,18 In addition, MRI can image bone marrow and soft tissues, detecting bone marrow oedema and structural abnormalities associated with diabetes. The Traditional Fracture Risk Assessment Tool (FRAX®) often underestimates the actual fracture risk in patients with diabetes, because they often have normal or higher BMD, yet have poor bone quality.19 However, taking into account specifically T2D, FRAXplus® identifies a statistically significantly higher 10-year risk of major osteoporotic fractures and hip fracture compared to risks calculated using the traditional FRAX® model.20 Therefore, this method may provide specific strategies to improve bone health and reduce the risk of fracture not only in postmenopausal women with diabetes, but also in individuals with IR.
Other clinical assessment tools used to evaluate the risk of secondary osteoporotic fractures include biochemical markers. Bone turnover markers not only enable us to assess the activity of osteoblasts and osteoclasts at a whole-body level, but also offer advantages such as being non-invasive, cost-effective, and allowing for multiple measurements over time.21 For example, C-terminal telopeptide (CTX) and procollagen Type I N-terminal propeptide (P1NP) provide insights into bone remodelling dynamics. Recent findings reported that markers of bone formation and bone resorption are lower in patients with T2D compared to those without T2D.8,21 Indeed, the results of a systematic review and meta-analysis encompassing 66 studies also confirm that T2D is characterised by a condition in which bone metabolism is reduced.22 However, low bone turnover marker levels in patients with T2D do not consistently predict the risk of fracture; therefore, they should not be interpreted as an indicator of low fracture risk in these patients. Meanwhile, a comprehensive meta-analysis conducted by Yang et al.23 in 2024, encompassing 14 carefully selected observational studies, has revealed consistent changes in bone metabolism markers in patients with T2D. A significant reduction of 15.2% in osteocalcin levels and 12.8% in P1NP levels, markers of bone formation, was observed when compared with an age-matched group of non-diabetic controls. In contrast, the 18.4% reduction in CTX, a marker of bone resorption, indicates a generally suppressed bone turnover rather than the high bone turnover profile traditionally associated with osteoporosis.
DISCUSSION
There are complex pathological links and clinical effects between T2D and osteoporosis; in addition to IR and chronic inflammation, bone formation is impaired via hyperglycaemia, advanced glycation end products, oxidative stress, and vascular dysfunction.24 The pathogenesis of diabetic osteoporosis is also related to blood glucose fluctuations. Blood glucose fluctuations can lead to oxidative stress, increased levels of advanced glycation end products and inflammatory mediators, reduced insulin secretion, decreased 1,25-dihydroxycholecalciferol, elevated parathyroid hormone levels, and the occurrence of oxidative stress and autophagy. These factors, in turn, inhibit the function of osteoblasts and osteoclasts, leading to osteoporosis and osteoporotic fractures.25 Even in individuals with normal or high BMD, inflammation-associated IR, characterised by a decline in bone quality, accelerated bone loss, and an increased risk of fractures, is the primary cause of diabetic bone disease. Although it may seem paradoxical, patients with T2D are at significantly higher risk of hip, spinal, and pelvic fractures, particularly due to the deterioration in bone quality associated with hyperinsulinaemia. In addition to diabetic bone disease, an increase in features associated with metabolic syndrome, such as abdominal obesity, may also indicate an increased risk of fractures linked to IR and chronic inflammation. This is associated with an increased risk of fractures through a complex interaction that impairs bone quality and alters the properties of bone tissue. 7,12
Studies have shown that insulin, which regulates blood glucose levels via the PI3K/protein kinase B (AKT) signalling pathway, promotes cell proliferation and osteogenic differentiation.26 Insulin acts on osteoblasts via membrane receptors, stimulating DNA synthesis, triggering cell proliferation, and promoting the synthesis of osteocalcin and collagen, which are essential precursors for bone formation.27 In addition, insulin facilitates the differentiation of osteoblasts and the maturation of the bone matrix by increasing the expression of the RUNX2 gene, one of the key regulators of bone metabolism.28 In the early stages of T2D, systemic IR and the accompanying compensatory hyperinsulinaemia can lead to an increase in bone mineralisation. However, as the disease progresses to more advanced stages, the function of the pancreas’ β cells declines, and a decrease in BMD is observed alongside reduced insulin secretion.26,29 Existing evidence indicates that IR plays an important clinical role in the development of osteoporosis, and an increasing number of studies have demonstrated a significant negative correlation between IR and BMD. As impaired insulin signalling can suppress bone formation by reducing osteoblast proliferation and collagen synthesis, and consequently lead to a decrease in BMD, tests such as the Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) and Triglyceride-Glucose Body Mass Index (TyG-BMI), which serve as markers of IR, are useful in clinical diagnosis.8 For example, the results of a cross-sectional study suggesting that the TyG-BMI may better represent metabolic factors closely associated with bone health compared to the TyG index have shown that higher TyG-BMI levels are inversely associated with the risk of osteoporosis in patients with T2D.30 In another study, it was reported that a high TyG-BMI is a strong and accessible predictor of osteoporosis, particularly in young men, smokers, and individuals with preserved kidney function.18 Therefore, its integration into clinical practice could enable targeted interventions in high-risk populations. For example, although the pathogenesis of postmenopausal osteoporosis is traditionally addressed from an endocrine perspective, it is largely exacerbated by the effect of chronic, low-grade inflammation. However, it is recognised that secondary osteoporosis, which is associated with symptoms of sarcopenia and frailty in older women, triggers a shift from the single-organ frailty paradigm to the systemic frailty paradigm.31
A decline in metabolic flexibility is a key factor triggering this process; as the ability to switch between lipid and glucose oxidation diminishes with ageing and menopause, IR and increased susceptibility to oxidative stress sustain inflammatory signalling.32 Furthermore, postmenopausal ectopic fat accumulation and changes in the endocrine function of adipose tissue also contribute to IR and systemic inflammation. This, in turn, may influence fat accumulation in the bone marrow and increase osteoimmune activation.8,33 On the other hand, according to Dupuis et al.,34 the association between IR and sclerostin (positive) and osteocalcin (negative), but not with fat mass parameters, suggests that IR plays a predominant role over fat mass in the regulation of osteokines. Therefore, clinically, IR and T2D in postmenopausal women are linked to elevated fragility‐fracture risk (often disproportionate to BMD), arguing for combined metabolic and skeletal risk stratification in postmenopausal osteoporosis management.1,14 On the other hand, dysbiosis, which refers to an imbalance in the gut microbiome, is one of the factors contributing to chronic, low-grade systemic inflammation. This condition accelerates bone resorption while suppressing bone formation, thus contributing to the development of osteoporosis.35 As an imbalance in the gut microbiota significantly contributes to secondary osteoporosis in T2D by triggering chronic, low-grade systemic inflammation and disrupting nutrient metabolism, restoring microbial balance through diet or targeted treatments has become a new focus in the management of diabetic bone disease.36
Insulin and insulin-like growth factor-1 (IGF-1) deficiencies drive diabetic osteoporosis primarily by creating an ‘anabolic deficit’ in bone remodelling. Their absence impairs osteoblast differentiation and collagen synthesis while dysregulating key signalling pathways (e.g., PI3K/AKT and Wnt/β-catenin) that are essential for building and maintaining bone mass.37 Furthermore, in diabetes, glucolipotoxicity, the combined metabolic stress resulting from high glucose and high lipid levels, is another factor contributing to the development of diabetic osteoporosis via the chronic activation of NLRP3 inflammasome. This excessive activation severely disrupts bone remodelling by halting bone formation and accelerating bone resorption. This leads to skeletal fragility and an increased risk of fractures.38 The potential contribution of IR and IGF-1 deficiency to the development of diabetic osteoporosis suggests that glucagon-like peptide-1 (GLP-1) receptor agonists may play a role in treatment or prevention. GLP-1 receptor agonists and dipeptidyl peptidase 4 (DPP-4) inhibitors indirectly improve bone metabolism by stimulating glucose-dependent insulin secretion. Through this incretin-driven pathway, GLP-1 triggers the physiological release of insulin, which is known to possess anabolic (bone-forming) and antiresorptive properties.39 As GLP-1 and glucose-dependent insulinotropic polypeptide receptors are also expressed in bone marrow stromal cells and osteoblasts, this implies that these hormones can stimulate bone formation entirely independently of their glucose-lowering effects. Similarly, DPP-4 inhibitors also indirectly improve bone metabolism by stimulating glucose-dependent insulin secretion. Whilst some clinical studies suggest that GLP-1 receptor agonists and DPP-4 inhibitors may reduce the risk of fractures, a meta-analysis has not found any significant association between the use of DPP-4 inhibitors and the risk of fractures.40,41 Indeed, experimental and clinical studies found that GLP-1 receptor agonists had a significant positive effect on bone quality and strength, possibly by improving the blood supply to the bone necessary for bone health.42,43 In addition, potential therapeutic advances in the management of bone-related diabetes include novel drug therapies aimed at improving bone density and strength; these include drugs that may offer targeted benefits, such as selective receptor activator of NFB ligand (RANKL) inhibitors and sclerostin antibodies.44
CONCLUSION
As a result, chronic systemic inflammation and IR act as a synergistic, bidirectional pathway in the development of secondary osteoporosis, creating a state of ‘metabolic bone disease’ that often exceeds the fracture risk of age-associated osteoporosis and postmenopausal osteoporosis. Chronic inflammation (e.g., from metabolic syndrome, diabetes, or autoimmune disease) induces IR, while IR fuels further inflammation, together driving accelerated bone loss via increased bone resorption and suppressed bone formation, then diabetic bone disease. Patients with T2D who have chronic inflammation and IR should be actively screened for bone health, as they are at high risk of undiagnosed secondary osteoporosis. To diagnose both diabetic bone disease and potential secondary osteoporosis, it is necessary to assess bone quality and integrity using imaging techniques and biomarkers. Understanding this co-existent causal relationship is important for developing new treatments for conditions that affect both diabetic bone diseases and secondary osteoporosis.