Speakers: Priya Kishnani,1 Mark Roberts,2 Stephan Wenninger,3 Annic Kolbrück,4 Staci Kallish5
1. Department of Pediatrics, Division of Medical Genetics, Duke University Medical Center, Durham, North Carolina, USA
2. Greater Manchester Neurosciences Unit, Salford Royal NHS Foundation Trust, Salford, UK
3. Ludwig Maximilian University of Munich, Germany
4. Person living with LOPD, Germany
5. Penn Medicine, Philadelphia, Pennsylvania, USA
Support: This is a promotional article, initiated and funded by Amicus and contains information on Amicus products. It is based on a promotional satellite symposia at the WORLD congress 2025, held in San Diego on February 4, 2025. Amicus funded medical writing assistance from Amanda Barrell, Brighton, UK, and provided editorial comment.
Disclaimer: This article contains discussion of cipaglucosidase alfa(▼) and miglustat. Cipaglucosidase alfa is a long-term enzyme replacement therapy used in combination with the enzyme stabiliser miglustat for the treatment of adults with late-onset Pompe disease (acid alpha-glucosidase [GAA] deficiency). This treatment is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See information box at the end of this article for how to report adverse reactions. Study results and indications presented during the symposium were consistent with the United States label for cipaglucosidase alfa + miglustat. Data in this summary article have been adapted to align with the European and UK Summary of Product Characteristics (SmPC) and indication.
HCPs in the European Union, see cipaglucosidase alfa full prescribing information here.
HCPs in the European Union, see miglustat full prescribing information here.
HCPs in the UK, see full cipaglucosidase alfa prescribing information here.
HCPs in the UK, see full miglustat prescribing information here.
Disclosure: Kishnani has received research/grant support from Sanofi Genzyme and Amicus Therapeutics, Inc.; consulting fees and honoraria from Sanofi Genzyme, Amicus Therapeutics, Inc., Bayer, and Asklepios Biopharmaceutical, Inc. (AskBio); is a member of the Pompe and Gaucher Disease Registry advisory board for Sanofi Genzyme, the Pompe disease advisory board for Amicus Therapeutics, Inc., and an advisory board for Baebies; and has held equity in Asklepios Biopharmaceuticals, Inc. and may receive milestone payments related to that equity in the future. Roberts has received fees for advisory boards and speaker honoraria from Sanofi, BioMarin, and Amicus Therapeutics, Inc. Wenninger has received a research grant from the Deutsche Gesellschaft für Muskelkranke eV (DGM); served on advisory boards for Alexion Pharma, ARGENX Therapeutics, Amicus Therapeutics, and Sanofi Genzyme GmbH; and has received funding for travel or speaker honoraria from Selpers og, Sanofi-Aventis Germany GmbH, SH Glykogenose Gesellschaft, Alexion Pharma GmbH, ARGENX Therapeutics, Amicus Therapeutics Inc., and Sanofi Genzyme GmbH. Kolbrück has no disclosures. Kallish has received grants and research support from Amicus Therapeutics, Inc., Astellas Pharma, Idorsia, Incyte, Ipsen Biopharmaceuticals and Sanofi Genzyme GmbH; and consulting fees from Amicus Therapeutics, Inc., and Sanofi Genzyme GmbH.
Keywords: Acid alpha-glucosidase (GAA), enzyme replacement therapy (ERT), glycogen storage disease Type II (GSDII), late-onset Pompe disease (LOPD), muscle weakness, Pompe disease (PD), respiratory function, treatment monitoring.
![]()
Meeting Summary
This symposium, ‘A case-based approach to navigating treatment switch in late-onset Pompe Disease (LOPD)’, was held at the 21st WORLDSymposium from the 3rd–7th February 2025, in San Diego, California, USA. Speakers discussed some of the unmet needs for patients living with LOPD, used clinical cases to highlight key considerations for navigating a treatment switch, and presented data on cipaglucosidase alfa + miglustat for enzyme replacement therapy (ERT)-experienced adults living with LOPD. LOPD is a lysosomal disorder caused by a deficiency of the acid alpha glucosidase (GAA) enzyme which leads to progressive glycogen accumulation in muscle. As a multisystemic, progressive, heterogeneous disease, LOPD can lead to irreversible muscle damage and weakness, reducing a person’s ability to move and breathe. The ERT alglucosidase alfa has been the cornerstone of LOPD management since 2006, but waning efficacy in some patients, typically after 3–5 years, creates a need for additional treatment options. Three key mechanistic challenges exist that could result in suboptimal delivery of enzymes to tissues: enzyme stability in the blood stream, enzyme uptake into the muscle cells, and enzyme activity in the lysosome. During the symposium, the speakers, experts in the management of LOPD, explored how second generation treatment options may help address current unmet needs, and how shared-decision making and holistic monitoring can support informed treatment decisions in certain adults with LOPD. They reviewed key data on cipaglucosidase alfa + miglustat for ERT-experienced adults living with LOPD, and shared their insights on the management of treatment switching using clinical cases.Introduction
LOPD is a progressive, multisystemic, lysosomal disorder which can lead to irreversible muscle damage and profoundly impact people’s lives.1 LOPD results from a deficiency of the GAA enzyme, which is responsible for the hydrolysis of glycogen to glucose in the lysosome. GAA deficiency impairs glycogen degradation, causing progressive glycogen accumulation within muscle tissue.1 It is heterogeneous, ranging from mild muscle involvement to severe chronic respiratory failure and marked limb-girdle and axial weakness.2 As the disease progresses, the lysosomes rupture, releasing glycogen and autophagic debris into the tissue, which can displace the elements that are vital for muscle contraction.3 This results in progressive muscle weakness that can reduce a person’s ability to breathe and move.3 “The end stage of muscle damage, where there is fatty replacement, really is a point of no return,” said Priya Kishnani, Department of Pediatrics, Division of Medical Genetics, Duke University Medical Center, Durham, North Carolina, USA, emphasising the importance of early diagnosis and timely treatment, “so that we can try to salvage muscle” and “bring patients to their healthiest”.
Kishnani highlighted the burden of LOPD with data from a Dutch study of 54 people with untreated disease. The mean age of symptom onset was 28 years, and first complaints were mostly related to mobility and limb-girdle weakness.
The mean age of diagnosis was 35 years, which was approximately the age that most people reported experiencing problems using stairs or standing up. Almost half (48%) of participants were using a wheelchair at the time of the study, with this becoming necessary at a mean age of 46 years. A total of 37% of people in the study were using ventilatory support, which was typically initiated around the age of 49 years.4 “The progression of skeletal and respiratory muscle weakness is associated with decreased quality of life and increased mortality,” she said.5
Unmet Needs
The first recombinant human GAA (rhGAA) ERT, alglucosidase alfa, became available in 2006.6,7 However, while alglucosidase alfa marked a major step forward for people living with the condition, it is also associated with limitations. Kishnani explained that after initial benefits, many patients experience worsening symptoms after 3–5 years on treatment, including decreased walking ability and pulmonary function, which indicates waning efficacy.8-10 For example, one study of 30 people with LOPD on algucosidase alfa found a 22.2 percentage point decrease from baseline of 49% predicted 6MWT, and a decrease of 11 points from baseline of 54% predicted FVC after 10 years. Other long-term studies of alglucosidase alfa show the same pattern of short-term improvements followed by waning efficacy,8-10 which highlights why additional treatments are needed, said Kishnani.
Three key mechanistic challenges with rhGAA delivery to target tissues exist,11,12 and only around 1% of enzyme is taken up into the skeletal muscle 24 hours after intravenous bolus administration.11 The first challenge is enzyme stability. rhGAA is most stable and active at the acidic pH of the lysosome, but it is delivered to the tissues via the bloodstream, which has a near-neutral pH. As such, the enzyme becomes unstable and inactivated.11 The second challenge relates to enzyme uptake. ERT relies on rhGAA being taken up from outside the cell, through the exogenous pathway using the cation-independent mannose 6-phosphate receptor (CI-MPR). However, rhGAA is poorly phosphorylated, which can limit uptake by target muscle cells. Non-target tissues, such as the liver, may clear the vast majority of ERT from systemic circulation, leading to low concentrations in the interstitial space.11 The third challenge is around enzyme activity. The enzyme must undergo intracellular proteolytic cleavage and N-glycan trimming to yield its most active form.12 “If we had more optimal delivery, we could clear more glycogen efficiently,” said Kishnani. “One way to achieve this would be with an enzyme that is more stable in the neutral pH of the bloodstream. The second is to increase the uptake, which can be done through more M6P or more bis-M6P, which has a much higher affinity than M6P. The third is by making sure the enzyme is actively degraded in the lysosome to its most active, mature form, so it can do its job.”
Cipaglucosidase alfa + miglustat, the first and only two-component therapy for adults with LOPD, was designed to address key challenges in delivering rhGAA, she explained (Figure 1). Cipaglucosidase alfa is naturally derived from a Chinese hamster ovary (CHO) cell line, resulting in cellularly derived (CHO) N-glycans. It is a bis-mannose-6-phosphate (M6P)-enriched enzyme, which has approximately 3,000 times higher binding affinity to the CI-MPR than that of M6P, and it is designed to increase uptake into muscle cells.13 Once inside the muscle cell, it can be processed into its most active form to break down glycogen. Cipaglucosidase alfa must be taken with miglustat, an oral enzyme stabiliser, which binds with and reduces the inactivation of cipaglucosidase alfa in the blood, increasing the amount of active enzyme available to target skeletal muscles.14
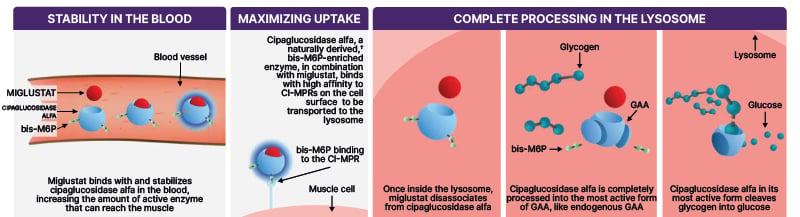
Figure 1: Cipaglucosidase alfa and miglustat is designed to address key challenges in delivering rhGAA.12-16*
*Based on in vitro data.
?Enzyme derived from a Chinese hamster ovary (CHO) cell line using perfusion methodology, resulting in cellularly derived (CHO) N-glycans..
bis-M6P: bisphosphorylated mannose 6-phosphate; CI-MPR: cation-independent mannose 6-phosphate receptor; GAA: acid a-glucosidase; rhGAA: recombinant human acid a-glucosidase.
Cipaglucosidase Alfa + Miglustat: Clinical Trial Data
Kishnani presented key clinical trial data for cipaglucosidase alfa + miglustat, from the Phase III PROPEL study. The highlighted results were consistent with the USA label for cipaglucosidase alfa + miglustat, though the data summarised below have been adapted to align with the European and UK Summary of Product Characteristics (SmPC) and indication.
PROPEL was a 52-week head-to-head, double-blind, active-controlled, international, multi-centre clinical study, in which 122 adults living with LOPD were randomised 2:1 to receive cipaglucosidase alfa + miglustat, with dosing based on the subject’s weight, or alglucosidase alfa in combination with placebo every other week. The primary endpoint was 6MWD change from baseline to Week 52 for comparison of superiority, while sitting percent predicted FVC was the secondary endpoint. Key pharmacodynamic endpoints included Hex-4 and creatine kinase (CK). While the overall study population included both ERT-experienced and ERT-naïve patients, most participants (n=95) had previously received alglucosidase alfa, with a mean treatment duration of more than 7 years before entering the trial.13
In the overall study population at week 52, there was a mean improvement, from baseline, of 20 m in 6MWD.13 Among those in the alglucosidase alfa + placebo group, the mean improvement from baseline was 8.3 m, indicating a cipaglucosidase alfa + miglustat treatment effect of 11.7 m (95% CI: -1.0, 24.4). However, the primary endpoint of superiority was not met (p=0.07). The mean change in FVC from baseline was -1.4% in the cipaglucosidase alfa + miglustat group, compared with -3.7% in the alglucosidase alfa + placebo group, indicating a cipaglucosidase alfa + miglustat treatment effect of 2.3% (95% CI: 0.2, 4.4).13
Among the ERT-experienced participants in the cipaglucosidase alfa + miglustat group (n=65), there was a mean improvement in 6MWD of 15.8 m from baseline to 52 weeks, compared with 0.9 m for the alglucosidase alfa + placebo group (n=30). This indicated a cipaglucosidase alfa + miglustat treatment effect of 14.9 m (95% CI: 1.2, 28.6). In terms of respiratory function, the ERT-experienced subjects in the cipaglucosidase alfa + miglustat group showed a mean FVC change of -0.2% from baseline to Week 52. For those in the alglucosidase alfa + placebo group, the change was -3.8%, indicating a treatment effect of 3.6% (95% CI: 1.3, 5.9). In those treated with cipaglucosidase alfa + miglustat, there was a mean reduction in urine glucose tetrasaccharide (Hex-4) of -31.5%, compared to a mean increase of 11% in those in the alglucosidase alfa + placebo over the course of the trial.13 Kishnani reiterated the data pointing to waning efficacy of alglucosidase alfa after 3–5 years.8-10 Based on these prior studies, she explained “you would not expect to see further benefit” in some patients.
The proportion of patients who experienced treatment-emergent adverse events and infusion-associated reactions (IARs) was similar between the cipaglucosidase alfa + miglustat and the alglucosidase alfa + placebo group. The most commonly reported IARs only attributable to cipaglucosidase alfa were dizziness (2.6%), flushing (2.0%), somnolence (2.0%), chest discomfort (1.3%), cough, (1.3%), infusion site swelling (1.3%), and pain (1.3%). Headache was very common (≥1/10).
Adverse reactions reported during cipaglucosidase alfa infusion or within 2 hours after completion of this infusion in PROPEL were: abdominal distension, chills, pyrexia, dizziness, dysgeusia, dyspnoea, pruritus, rash, and flushing. Most adverse reactions were mild or moderate in severity and transient in nature. Reported serious adverse reactions only attributable to cipaglucosidase alfa were pharyngeal oedema (0.7%), presyncope (0.7%), pyrexia (0.7%), chills (0.7%), dyspnoea (0.7%), and wheezing (0.7%). A total of 0.7% of patients receiving cipaglucosidase alfa + miglustat experienced a serious adverse reaction of anaphylaxis during PROPEL, and 1.3% of patients receiving cipaglucosidase alfa + miglustat discontinued treatment due to adverse reactions.13
Regular, Holistic Monitoring
With the emergence of additional treatment options, healthcare professionals need to understand if and when to initiate treatment switch. Such decisions should, argued the speakers, be guided by holistic and routine monitoring.
This can be challenging, as “there is no one clinical test that shows us whether a patient is stable on a treatment,” said Stephan Wenninger, Ludwig Maximilian University of Munich, Germany. Validated monitoring assessments cover metrics across motor function, muscle strength, respiratory function, patient-reported outcomes (PRO), safety, and additional assessments, and their recommended use varies across guidelines.17-22 “Yet no patient is willing or able to do more than three or four of these,” Wenninger added.
To help address this challenge, Wenninger presented the recommendations of the European Pompe Consortium (EPOC), who defined a core data set that should be done in every patient to determine whether they are stable or would benefit from switching. This ‘start, switch, and stop’ (triple‐S) criteria for ERT (Figure 2) includes the 6MWT, manual muscle testing, seated/supine FVC, and the Rasch-built Pompe-specific Activity scale (R-Pact).23 The EPOC guideline recommends regular monitoring and considering the results together with patient experience to guide shared treatment decisions.23
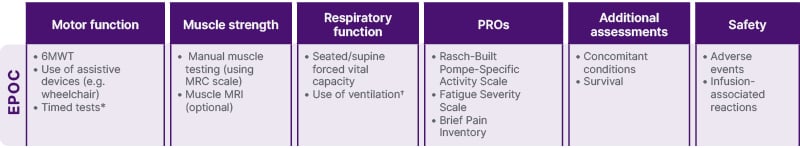
Figure 2: European Pompe Consortium-recommended assessments in late-onset Pompe disease.23
*E.g. time to climb four steps and time to get up from the floor.
†Non-invasive/invasive and number of hours.
6MWT: 6-minute walk test; MRC: Medical Research Council; PRO: patient reported outcome.
Moving from theoretical considerations to the real-world, Mark Roberts, Greater Manchester Neurosciences Unit, Salford Royal NHS Foundation Trust, Salford, UK, shared his experience of monitoring assessments from the UK Early Access to Medicines Scheme (EAMS). The EAMS is a pathway that permits switching from standard-of-care treatments to therapies pending authorisation in cases of clear unmet need. Thirty-four patients who initiated cipaglucosidase alfa + miglustat treatment at six specialist Pompe disease centres in England and Wales and consented to bi-annual study visits from enrolment (December 2021–July 2023) until January 2024. “This (registry) is now one of the largest cohorts in Europe of patients treated with cipaglucosidase + miglustat,” said Roberts. 6MWD, sitting FVC, and creatine kinase (CK) were recorded at all six study centres, but the frequency of other recommended assessments, such as supine FVC and urinary Hex-4, which is a breakdown product of glycogen, was variable.24 Roberts also revealed that one additional symptom the EAMS Registry was keen to examine was fatigue, which is a “huge problem for patients with Pompe”. He described how the Fatigue Severity Scale records the patient’s perception of fatigue, “so speaking to that patient-reported outcome that we all recognise as one of the most important metrics of all”.
Guiding Switching Decisions
When it comes to switching, Roberts said that “the first question in clinic” should always be: “Are they really stable?” Many patients, he added, will be deteriorating. “In terms of the metrics, I think it has to be holistic. It may be fatigue, it may be exercise intolerance, it may be falls, patient-reported outcomes (PRO), and, of course, respiratory function, timed walking, and so on are very important,” he said. If things are changing, it is an opportunity to have a “straightforward discussion” about switching. At his centre, patients who are switching treatments are seen at least twice a year, he added.
A focus on respiratory function, in particular, can be useful in detecting progression, said Wenninger. Hypoventilation syndrome, primarily due to respiratory muscle weakness, is common in neuromuscular disorders such as LOPD.
It is an important cause of morbidity and mortality, and its symptoms can be linked to disease progression.25 “We have to figure out whether their symptoms are attributed to respiratory function or to core symptoms of the neuromuscular disease,” he said. To detect respiratory decline, he recommended a suite of standard assessments, including FVC in the supine position, manometry (maximum inspiratory and expiratory pressures), peak cough flow and, where these tests are normal and the patient still has symptoms, “you have to add polysomnography”.26 One test, Wenninger said, “is not enough”. While he believes the tests should be performed “at least once a year”,23 in patients experiencing severe decline, scheduling assessments every 6, or even 3, months, would be more appropriate, he added.
The availability of additional LOPD therapies provides some patients with the opportunity for a second treatment switch, said Staci Kallish, Penn Medicine, Philadelphia, Pennsylvania, USA (Figure 3). She explained that the factors that inform her decisions around a second switch “are very similar to those that we think about when we make a first switch”. She also agreed with other speakers that, while monitoring assessments and immune reactions to treatment are important, clinicians also need to considerpatient preference: “People with LOPD tend to be very well informed. There are really strong patient communities, where patients are talking and learning from each other, and they may request a switch,” she said.

Figure 3: Considerations for a second treatment switch in adults with late-onset Pompe disease.
Expert opinion of the speaker.
Of course, clinical trial results remain the gold standard source of evidence during treatment decisions. Yet, Kallish noted, there are no head-to-head comparative data of the second generation LOPD treatments. This, she said, makes “our real-world experience even more important”.
Treatment Switch Case Studies
A key objective of this symposium was to share clinical experience of patients who had switched treatments, highlighting the considerations involved in guiding treatment choice. Roberts described three cases from the UK EAMS registry where patients switched from alglucosidase alfa to cipaglucosidase alfa + miglustat, noting that it was important to capture patient-reported benefits, as well as quantitative metrics.
The first case was a 65-year-old man who was diagnosed with LOPD in 2007 at the age of 48 years, having previously been diagnosed with limb-girdle dystrophy (Roberts: Data on file). This patient initiated alglucosidase alfa the following year and reported initial improvements; however, by 2022 he felt that the condition was progressing and, after 14 years on ERT, asked to enter EAMS and start on cipaglucosidase alfa + miglustat. Roberts described how this patient’s 6MWD and FVC were generally stable over the following 24 months and the patient reported exercising more frequently.
A second patient was a 47-year-old man who was diagnosed with LOPD at the age of 19 years, following family screening (Roberts, Data on file), and initiated alglucosidase alfa in 2007, at the age of 29 years. This patient experienced gradual disease progression between 2008–2022, including falls, which, due to a low BMI, put him at risk of fractures, although he was still independently mobile at this point. In 2022, at the age of 45 years and after 15 years on ERT, the patient asked to enter EAMS and was put on cipaglucosidase alfa + miglustat. Over the following 24 months, his 6MWD and FVC remained generally stable and the patient reported he had experienced fewer falls.
The third patient, a 59-year-old male, first noticed shortness of breath during exercise in 2000, at the age of 35 years (Roberts, Data on file). In 2012, he developed nocturnal hypoventilation and started on bilevel positive airway pressure ventilation before LOPD was diagnosed. The following year, at 48-years-old, he initiated alglucosidase alfa therapy. Symptoms initially improved, but by 2017 the patient felt the disease was progressing. He told his health team that he was getting to the stage where weakness was significantly impacting his day-to-day life, and his reduced mobility was a real concern. This gradual deterioration continued until 2022, when, after 9 years on ERT, he entered the EAMS and was initiated on cipaglucosidase alfa + miglustat. Since switching, his 6MWD and FVC have remained stable. Asked about his perception of treatment post-switch, the patient said he felt his strength was slightly worse, but he had more energy.
Kallish shared her insights on how to navigate cases involving a second treatment switch, noting that some patients may experience IARs with one therapy, and thus may benefit from a second treatment option. Her first patient case was a 61-year-old woman who was diagnosed in 2016, when she was 53 years old (Kallish, Data on file). This patient switched from alglucosidase alfa to avalglucosidase alfa in February 2023, but in November reported feelings of exhaustion since the early summer, and was concerned this was due to the treatment switch. In February 2024, she started on cipaglucosidase alfa + miglustat, yet experienced IARs from her second dose. These were unable to be managed by pre-infusion medications and the patient successfully switched back to avalglucosidase alfa in April.
Kallish’s second patient case was a 68-year-old man who developed chronic back pain in 2008, at the age of 52 years (Kallish, Data on file). Following a continual worsening of symptoms over time, he was diagnosed with LOPD in 2019, when he was 63 years old. This patient experienced IARs with alglucosidase alfa, which were managed using pre-infusion medications. After 3 years, when avalglucosidase alfa became available he was started on it, continuing the same pre-infusion medications. In January 2023, however, he developed an IAR that required the infusion to be stopped, and symptoms lasted until the next day. He switched back to alglucosidase alfa, but was concerned about disease progression, while Kallish was concerned about the risks of restarting avalglucosidase alfa in him. In December 2023, after cipaglucosidase alfa + miglustat became available, he agreed to try a second switch. “He was a great candidate,” said Kallish. “I was able to transition him to this therapy, and he has tolerated it well and remains on it.” Since then, he reports improved energy levels and fewer limitations in his daily activities.
Shared-Decision Making
The panel were aligned on the importance of shared decision-making for informed management decisions.27,28 Indeed, the EPOC guidelines state that switching should be discussed on a patient‐by‐patient basis.23 “When we see a severe decline in one or more assessments, we should ask whether it is having an impact on their daily activities,” said Wenninger. “We need to ask how the patient feels with their current treatment, and whether they feel there is a worsening of their symptoms.” HCPs should discuss all measurements and outcomes with the patient, and explain available treatments with the help of decision aids such as leaflets, he said.
Annic Kolbrück, who was diagnosed with LOPD in 2004, shared her own experiences of switching treatments. She said she “didn’t hesitate” to start on ERT in 2006, but after initial symptom stabilisation she started to experience a decline. “I was getting more pain, I was fatigued, and I couldn’t be on my feet so long anymore. All the natural things you like to do were getting really, really hard,” she explained, adding: “I was really seeking something else that could help me.” The switching process involved “a lot of discussion” with her healthcare provider as well as her own research, which included using information from patient advocacy groups. The switch to cipaglucosidase alfa + miglustat, she went on, had worked well for her.
Kolbrück’s advice to HCPs was to keep their explanations clear and simple, highlighting what patients can expect from a new treatment and what the differences between treatments are. She added that it is about helping people to overcome any fear of change, such as explaining how the four-hour fasting that is required with cipaglucosidase alfa + miglustat can fit in their lives.
It is a big decision for any patient, said Roberts, adding that his team works hard to ensure people are given the time and education they need. “We have a big discussion with the patients, then we contact them the following week to start to understand what they would want,” he said. The team utilise information leaflets developed by the UK’s Pompe Support Network, which are written in plain English. “We can then try to help them understand what choice that they want to make. If they want to, we bring them back again for another face-to-face discussion.”
Summary and Future Directions
Summing up, Kishnani said it was clear that “not one glove fits all”. The evolving LOPD treatment landscape may provide certain patients with the option to switch, making it “a very exciting time” in the field of lysosomal diseases, specifically Pompe disease. “Imagine going from where we started with no therapies, to the first ERT in 2006, to now, where we have three therapies available,” she said, adding that more are in the pipeline.
Switching decisions should be supported by holistic monitoring across multiple assessments, patient-specific shared-decision making, and evaluation of both clinical trial data and real-world experience for the available treatment options. In the future, earlier diagnosis and timely treatment, Kishnani hoped, could help prevent irreversible muscle damage and stop people arriving at “the point of no return.”
| Adverse events should be reported.
To report an adverse event in the UK (including Northern Ireland), reporting forms and information can be found at yellowcard.mhra.gov.uk. Adverse events should also be reported to Amicus on 0808 234 6864 or via email to [email protected]. To report an adverse event in Ireland, visit the Pharmacovigilance Unit at the Health Products Regulatory Authority (HPRA) at www.hpra.ie. Adverse events in Ireland should also be reported to Amicus on 1800 936 230 or via email to [email protected]. |
PP-AT-ALL-0002-0325
May 2025






