
INTRODUCTION
The genomic landscape of Hodgkin lymphoma (HL) is not well understood because of the scarcity of Hodgkin-Reed–Sternberg cells, which makes conventional genotyping difficult. Liquid biopsies have shown to be a feasible tool to unravel the genomic landscape of HL because Hodgkin-Reed–Sternberg cells release substantial amounts of circulating tumour DNA (ctDNA) into the blood.1-3
Although current treatment outcomes with staging, risk-factor-based, and/or PET-stratified treatment are excellent, interim PET is far from perfect and patients are often over-treated.4-5 Hence, the aim of this study was to understand the genomic landscape of HL using whole exome sequencing (WES) of liquid biopsies. With this approach, biological upfront risk stratification to ultimately individualise patient treatment, while also minimising side effects, was investigated.
METHODS
75 unselected, newly diagnosed, advanced-stage HL patients were included. All samples underwent pre-analytical processing, extraction of the ctDNA, and matching germline controls and high-depth WES (targeting 37 Mb of the human genome). An in-house customised pipeline was used for two-fold error reduction using unique molecular identifiers and digital error suppression. As part of the validation, results were compared to deep targeted sequencing (>2,000x coverage) and a per-patient concordance of 75% (95% confidence interval: 65%–86%) was found. Furthermore, allele frequencies by WES were highly correlated with the allele frequencies of targeted deep sequencing of ctDNA (r=0.8664; p<0.00001).
RESULTS
Several already known but also novel, recurrently mutated tumour-suppressor or oncogenes were identified. Frequent occurrence of previously described alterations in SOCS1, GNA13, IGLL5, TNFAIP3, and STAT6 were confirmed. In addition, novel recurrently mutated loci were identified in PRDM9, SIRPB1, and FLG2 (Figure 1).
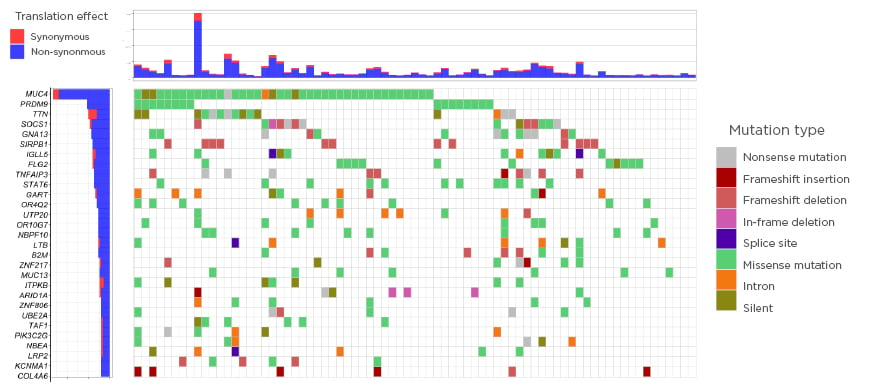
Figure 1: The genomic landscape in Hodgkin lymphoma.
Only significant tumour suppressor genes or oncogenes included (20/20 rule by Vogelstein et al.).6
Analysing significant copy number variations, recurrent deletions with peaks at 1p36.13 (SPEN), 6q23.3 (TNFAIP3), 14q13.2 (NFKBIA), 21q22.12 (RUNX1), and amplifications with peaks at 4q24 (NFKB1), 5p15,33 (TERT), 7q36.1 (MLL3), and 9p24.1 (PD-L1) were found.
When investigating COSMIC single-base substitution signatures (SBS), the most commonly identified was SBS1 (ageing), followed by SBS3 (failure of double-strand repair) and SBS9 (noncanonical AID). Interestingly, SBS25 (HL cell-line specific), previously only identified in HL cell-lines, was described in a patient cohort for the first time.
Several novel relationships within the genomic landscape of HL were identified and a subgroup with higher mutational burden associated with either mutations in B2M (p=0.0181) or high-level amplification of the PD-L1 locus (p=0.0031) was discovered. One might speculate that this subgroup of highly mutated HL relies on multiple immune-escape mechanisms.
By using non-negative matrix factorisation, two clusters of patients that shared similar genomic characteristics with distinct mutational drivers and clinical features were identified. Cluster 1 patients had higher Eastern Cooperative Oncology Group (ECOG) grades, whereas Cluster 2 patients had a higher proportion of nodular sclerosis, extra-nodal disease, and higher mutational burden.
Finally, a preliminary, biological risk stratification model based on high- and low-risk genetic alterations was created. A good outcome was defined as PET2-negativity, whereas PET-2-positivity was defined as a bad outcome. It was possible to distinguish three risk groups with the model. In the low-risk group, 8% of patients were PET-2-positive; this was 30% in the intermediate-risk group and 72% in the high-risk group.
CONCLUSION
In summary, this study is one of the most comprehensive studies looking at the genomic drivers of HL with a novel liquid biopsy approach.





