Meeting Summary
This symposium took place during the European Society of Intensive Care Medicine (ESICM) LIVES Congress 2025 in Munich, Germany. The chairperson for the symposium was Dan Longrois from Hôpital Bichat-Claude-Bernard, Assistance Publique-Hôpitaux de Paris, France. The aim of the symposium was to reframe vasoplegia by covering topics ranging from pathophysiology, clinical identification, and management to targeted therapy in the ICU. Firstly, Jean-Louis Vincent from the University of Brussels, Belgium, discussed the pathophysiology and diagnosis of vasoplegia, including a review of the main components of the renin-angiotensin-aldosterone system (RAAS), and the potential modulation of the RAAS with angiotensin II. Next, Gianluca Paternoster from the University of Basilicata and San Carlo Hospital, Potenza, Italy, described the clinical identification of vasoplegia and the challenges associated with managing patients with refractory vasoplegic shock. Following this, Ricard Ferrer from Vall d’Hebron University Hospital, Barcelona, Spain, described angiotensin II in vasoplegic shock and the potential for a targeted therapeutic option. Finally, Emily See from The Royal Melbourne Hospital, University of Melbourne, and the Australia and New Zealand Intensive Care Research Centre, Monash University, Melbourne, Australia, discussed applying angiotensin II in clinical practice, using evidence from clinical experience, clinical trials, and real-world practice.
Understanding Vasoplegia: From Pathophysiology to Diagnosis
Jean-Louis Vincent
Vasoplegia is defined as sustained low systemic vascular resistance in the context of a normal or high cardiac index, leading to uncontrolled vasodilation and reduced blood pressure.1,2 Vincent explained that arterial pressure is determined by blood flow (i.e., cardiac output) and vascular tone,3 and that hypotension is not necessarily caused by vasoplegia. He emphasised that in cases of septic shock, a comprehensive haemodynamic assessment, including blood flow and measurement of systemic vascular resistance, is needed before a definitive diagnosis of vasoplegia can be made.4
The activation of the RAAS involves angiotensinogen, which is transformed into angiotensin I (catalysed by renin) and then into angiotensin II (catalysed by angiotensin-converting enzyme [ACE]; Figure 1).5 Vincent explained that levels of ACE1 are decreased in the context of endothelial cell dysfunction and in the presence of substances counteracting ACE (ACE inhibitors; Figure 1). The angiotensin 1 and 2 receptors (AT1 and AT2 receptors) are important components of the system, with the AT1 receptor primarily leading to vasoconstriction and stimulating the release of aldosterone.6
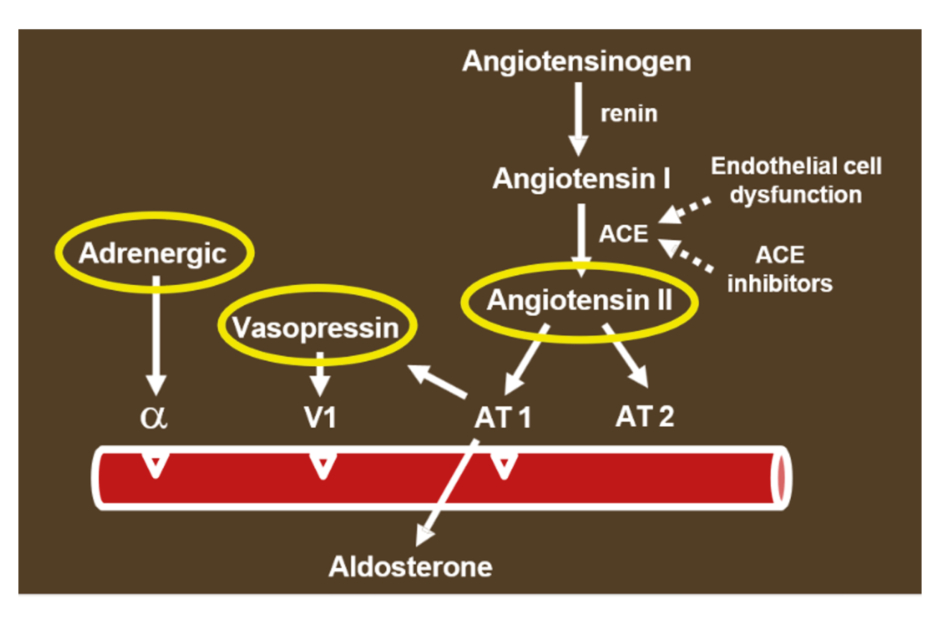
Figure 1: Activation of the renin-angiotensin-aldosterone system.
α: α-adrenergic receptor; ACE: angiotensin-converting enzyme; AT1: angiotensin 1 receptor; AT2: angiotensin 2 receptor; V1: vasopressin receptor 1.
There are three primary methods to increase blood pressure: α-adrenergic stimulation, vasopressin, and angiotensin II.3,7,8
Angiotensin II was first used in shock states in 1961, when there were reports of a vasopressor effect that could increase blood pressure.9-11 Modulation of the RAAS with angiotensin II was proposed as a new therapeutic intervention in sepsis in 2010.12 The ATHOS-3 trial, published in 2017, showed that angiotensin II administration increased blood pressure in patients with vasodilatory shock who did not respond to high doses of conventional vasopressors.13
Angiotensin II exerts several effects beyond vasoconstriction.14 Therefore, decreased angiotensin II levels may have consequences beyond inadequately low vascular tone.
The transformation of angiotensin I to angiotensin II is impaired in distributive shock, with an increased angiotensin I/angiotensin II ratio associated with higher vasopressor requirements and higher mortality rates.15
A recent report showed that in patients with septic shock, there is an increased angiotensin I/angiotensin II ratio and decreased circulating ACE activity compared with healthy controls.16 Vincent noted that in this scenario, there must be an increase in renin levels via a feedback mechanism.14 Indeed, the induction of sepsis in a non-clinical septic shock model resulted in a significant increase in plasma renin activity (p<0.0001).17
In a heterogeneous ICU population in which not all patients were in shock, serum renin levels were a marker of tissue perfusion and a more reliable predictor of ICU mortality than lactate levels.18 Serum renin levels were consistently reported to be related to the degree of renal dysfunction and survival in critically ill patients, with high renin levels associated with increased mortality risk.19
The data from the ATHOS-3 study showed that the administration of angiotensin II for catecholamine-resistant vasodilatory shock led to decreased renin levels.20 Patients with renin levels above the study population median (hazard ratio [HR]: 0.56; 95% CI: 0.35–0.88; p=0.01) appeared to derive survival benefit from angiotensin II administration compared to those with renin levels below the median (HR: 1.11; 95% CI: 0.66–1.86; p=0.70).20
In an experimental hyperdynamic sepsis model, mean arterial pressure (MAP) was higher with angiotensin II compared with placebo, whereas cardiac output and renal blood flow were lower compared with placebo.21 Despite this, urine output and creatinine clearance were higher in the angiotensin II group compared with the placebo group.21 Vincent underlined that even though the renal blood flow was decreased, there was an improvement in renal function.21
ACE2 leads to the production of angiotensin 1–9 and 1–7, which decreases the effect of angiotensin II on the vasculature.14 Vincent noted that non-clinical studies indicate that the early administration of angiotensin 1–7 may prevent the development of septic shock.22
However, to complicate matters, the enzyme dipeptidyl peptidase 3 (DPP3) hydrolyses angiotensin II, and high levels of circulating DPP3 are associated with worse outcomes in sepsis.14,16
Angiotensin II has vascular, renal, pulmonary, and immunological effects.14 Treatment with angiotensin II may reduce the incidence of organ failure and improve outcomes in patients with vasodilatory shock and sepsis.14 The response to angiotensin II differs between patients, with responsiveness considered a good prognostic sign.23 Vincent indicated that patients who might benefit most from angiotensin II are those with a confirmed hyperkinetic state (high cardiac output), renal dysfunction or failure,24 recent exposure to ACE inhibitors,25 or elevated blood renin levels,20,26 although Vincent acknowledged that renin levels are currently difficult to measure. Other factors, such as cardiac surgery, high requirements for norepinephrine, and acute respiratory failure, may also be important indicators for patients who might benefit from angiotensin II.27
Vincent summarised that the dysregulation of the RAAS in sepsis is a complex phenomenon; however, the clinical modulation of the RAAS in critically ill patients is promising and cannot be ignored.14,28,29
Clinical Identification and Management Challenges
Gianluca Paternoster
Paternoster specified that distributive shock is due to severe peripheral vasodilation and is common in patients with sepsis or following cardiopulmonary bypass (CPB). The hypotension that develops in sepsis is due to many factors, including RAAS imbalance or dysregulation, noradrenergic system dysregulation, and decreased response to norepinephrine, vasopressin, and angiotensin II.30 Key factors in the development of hypotension following CPB include increased synthesis of nitric oxide, reinfusion of cell saver blood, systemic reperfusion, and generation of oxygen free radicals.31 Paternoster clarified that the aim of therapy for sepsis or CPB is to restore systemic vascular resistance. Refractory vasoplegic shock is defined as an impaired responsiveness to vasopressor medications.32
Vasoplegia is defined as reduced systemic vascular resistance, with inadequate response to standard vasopressors, in the context of preserved or high cardiac output.1,2 Paternoster explained that a reduction in systemic vascular resistance leads to juxtaglomerular regulation with opposite effects on renin.33 A decrease in sodium intake stimulates the synthesis and release of renin, whereas an increase in sodium intake inhibits the synthesis and release of renin, which restores systemic vascular resistance.33
Under normal physiological conditions, the RAAS is balanced with regard to vasoconstriction versus vasodilatation, and pro-fibrotic versus anti-fibrotic effects.34 The RAAS is unbalanced during sepsis and following CPB, when there is increased vasoconstriction and fibrosis.34
Paternoster noted that the upregulation of AT2 receptors, and the additional presence of downregulation of AT1 receptors (i.e., the unbalanced regulation of both types of receptors, with AT2 receptors much more represented than AT1 receptors) leads to vasodilation and refractory vasoplegia.35
Paternoster explained that AT1 receptor downregulation blunts catecholamine sensitivity; therefore, RAAS dysfunction is associated with a higher norepinephrine requirement.36 According to Paternoster, norepinephrine is usually given first-line at a starting dose of 0.05–0.1 μg/kg/min intravenous (IV). The dose of norepinephrine is titrated to maintain a MAP of ≥65 mmHg; however, high doses of catecholamines are associated with increased risk of arrhythmia, myocardial toxicity, and receptor desensitisation.36
The absence of pulsatile flow during CPB exacerbates the process of reduced sodium intake. In a recent study, patients who did not have vasoplegia had an instantaneous and strong RAAS response after CPB, as shown by high levels of renin and angiotensin up to 48 hours after CPB, which means that the RAAS is balanced and working effectively.37 In contrast, patients with vasoplegia had noticeably diminished renin and angiotensin II responses.37
Although the use of renin as a predictive biomarker for angiotensin II responsiveness is biologically plausible,38 Paternoster questioned whether renin is the most appropriate parameter to measure.26
Paternoster explained that there is a strong relationship between the brain renin-angiotensin system, the sympathetic nervous system, and the secretion of catecholamines. However, if there is dysfunction in the renin-angiotensin system, there is simultaneous dysfunction in the sympathetic nervous system, which reduces the secretion of norepinephrine and epinephrine, because the adrenergic system is not working.
In this case, according to Paternoster, a non-adrenergic approach may improve outcomes in patients with refractory vasoplegic shock during sepsis39 or following CPB. This approach has proved successful in patients with septic shock, following cardiac surgery, and after non-cardiac surgery.39 A pathophysiological non-adrenergic approach might be effective during liver or renal transplantation.28
Expert consensus statements have been published that identify which patients might benefit most from angiotensin II for distributive shock, the potential barriers to the use of angiotensin II, and the gaps in the evidence.40
Paternoster summarised that patients with refractory vasoplegia can be identified clinically by considering the following parameters: RAAS dysfunction, blunted renin and angiotensin II response, reduced ACE1, increased ACE2, dysfunction of other angiotensin II effectors, and attenuated response to aldosterone and vasopressin.
Paternoster concluded, “I think it is time to move away from a strong physiological approach and consider a non-adrenergic approach to rebalance the RAAS in patients with refractory vasoplegia.”
Angiotensin II in Vasoplegic Shock: A Targeted Therapeutic Option
Ricard Ferrer
Ferrer remarked that evidence to support the rationale for the administration of external angiotensin II in patients with sepsis and vasodilatory shock is provided by non-clinical studies. In a porcine model of faecal peritonitis, angiotensin II reversed hypotension caused by sepsis and produced systemic and regional haemodynamic effects comparable to those observed with norepinephrine.41 Both angiotensin II and norepinephrine had a vasoconstrictor effect and restored MAP and mean pulmonary arterial pressure.41
In a rat model of sepsis-induced systemic haemodynamic dysfunction, changes to systemic haemodynamics, microcirculatory deterioration, and renal damage were ameliorated by angiotensin II.42 Increased urine output and some protection of the lipopolysaccharide-induced histological damage were observed with angiotensin II.42
In a rabbit study, intrarenal angiotensin II produced a selective decrease in afferent versus efferent arteriole diameter; however, efferent resistance was greater than afferent due to smaller resting luminal dimensions.43 In addition, in a sheep model of Escherichia coli-induced sepsis, urine output and creatinine clearance were greater with angiotensin II compared with placebo.21
A systematic review revealed that higher serum creatinine, lower urine output, and increased requirements for renal replacement therapy (RRT) were observed with catecholamines compared with non-catecholamine vasopressors in clinical studies of vasodilatory shock. In addition, there was an increase in RRT liberation in patients on high-dose catecholamines who received adjunctive angiotensin II.44
In the ATHOS-3 study, patients with vasoplegia receiving high doses of vasopressors (>0.2 μg/kg/min norepinephrine-equivalent dose [NED]) were randomised to angiotensin II or placebo.13 The primary efficacy endpoint was a response with respect to MAP at 3 hours after the start of infusion. This endpoint was reached by 69.9% of patients who received angiotensin II and 23.4% who received placebo (odds ratio [OR]: 7.95; 95% CI: 4.76–13.3; p<0.001).13
The dose of catecholamines was decreased for patients who received angiotensin II and increased for those who received placebo (mean change in NED from baseline to 3 hours: -0.03±0.10 and 0.03±0.23, respectively; p<0.001).13
There was also a trend for reduction in all-cause mortality for angiotensin II versus placebo (Day 28; HR: 0.78; 95% CI: 0.57–1.07; p=0.12), and angiotensin II had a manageable safety profile.13
Endothelial injury during shock can lead to ACE defects, which may cause decreased production of angiotensin II and increased renin (hyperreninemia).45 Ferrer indicated that patients with hyperreninemia might benefit from exogenous angiotensin II.20,26
Post-hoc analysis in the ATHOS-3 study showed that patients with hyperreninemia who received angiotensin II had decreased angiotensin I and renin, confirming that the angiotensin I-renin axis was dysfunctional and the administration of angiotensin II was correcting it.20 As mentioned by Vincent, in patients with renin concentrations above the study population median, angiotensin II significantly reduced 28-day mortality compared with placebo (unstratified HR: 0.56; 95% CI: 0.35–0.88; p=0.01; Figure 2A).20 A survival benefit was not seen in those with renin concentrations below the study population median (HR: 1.11; 95% CI: 0.66–1.86; p=0.70; Figure 2B).20
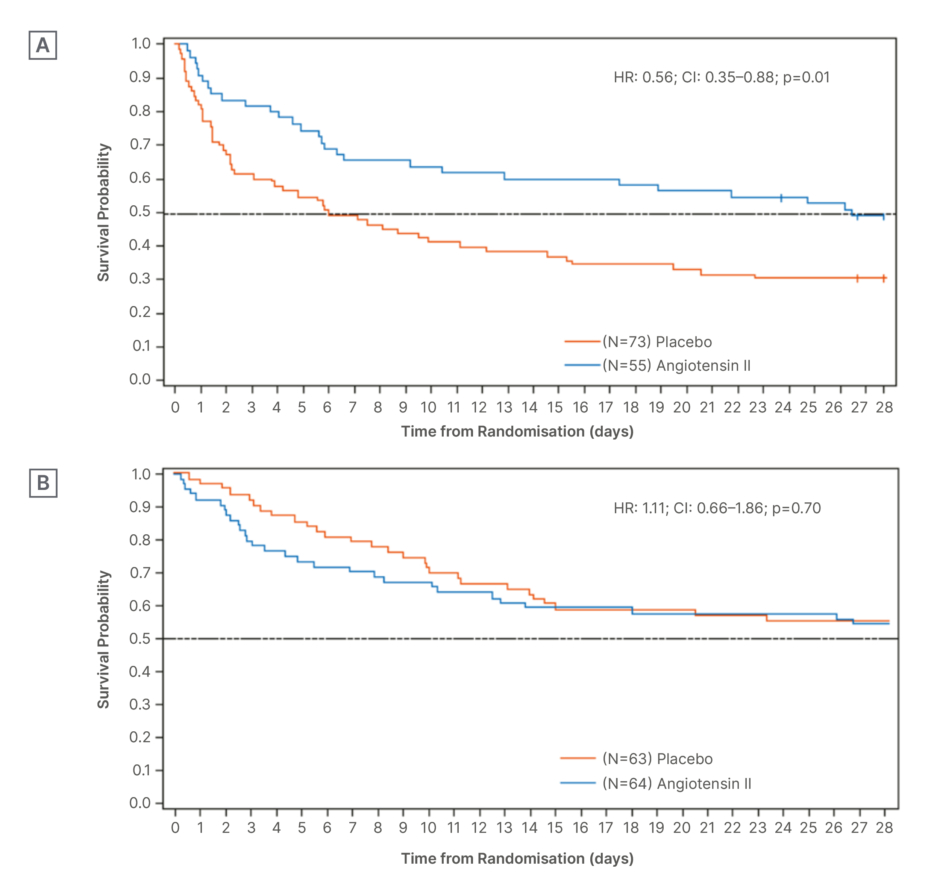
Figure 2: Kaplan-Meier survival plot according to renin concentrations and treatment with angiotensin II
or placebo.20
A) Day 28 survival: renin concentration above population median.
B) Day 28 survival: renin concentration below population median.
HR: hazard ratio.
Patients with prior exposure to ACE inhibitors in the ATHOS-3 study had increased cardiovascular response to angiotensin II and a greater decrease in NED compared with patients with no exposure.25 Ferrer indicated that patients with prior exposure to ACE inhibitors are good candidates for angiotensin II therapy.25 In contrast, there was a decreased cardiovascular response to angiotensin II in patients with previous exposure to angiotensin receptor blockers and little change in NED.25
The ATHOS-3 study indicated that patients on a lower NED at inclusion benefit from a secondary vasopressor.46 After adjusting for severity of illness, patients randomised to angiotensin II in the ≤0.25 µg/mg/min NED subgroup were half as likely to die at 28 days compared with those on placebo (HR: 0.509; 95% CI: 0.274–0.945; p=0.03).46 There were no differences in 28-day survival between the angiotensin II and placebo groups in the >0.25 µg/mg/min NED subgroup (HR: 0.933; 95% CI: 0.644–1.350; p=0.71).46
Ferrer commented that early administration of angiotensin II might be particularly beneficial.27,46 Post-approval data47 are concordant with those from the ATHOS-3 study.
Post-hoc analysis of ATHOS-3 study data showed that patients with acute kidney injury (AKI) in the angiotensin II group had lower 28-day mortality versus the placebo group (unadjusted HR: 0.52; 95% CI: 0.30–0.87; p=0.012), and were more likely to discontinue RRT within 7 days (adjusted HR: 2.90; 95% CI: 1.29–6.52; p=0.007), indicating another patient population potentially suitable for angiotensin II treatment.24
There is an association between ACE genetic polymorphism and risk of acute respiratory distress syndrome (ARDS), particularly in patients with sepsis.48 Patients with ARDS in the ATHOS-3 study who received angiotensin II had improved oxygenation; this was not seen in patients receiving a placebo.49
In the ATHOS-3 study, 16 patients had vasoplegia following CPB, and 88.9% of these patients responded to angiotensin II versus 0% on placebo (p=0.0021).50
An RCT investigating angiotensin II or norepinephrine (cited as noradrenaline) as a primary vasopressor for patients with vasoplegia following cardiac surgery showed that patients who received angiotensin II had shorter duration of vasopressors, lower incidence of AKI, and were more likely to achieve the target MAP (70–80 mmHg).51
Ferrer concluded that patients with distributive shock who are fluid resuscitated, receiving vasopressor therapy (0.2–0.5 µg/kg/min NED), with persistent hypotension, and no signs of cardiac dysfunction, are the target population for angiotensin II.27 Patients with distributive shock who have hyperreninemia, low-dose vasopressor, AKI requiring RRT, or sepsis-associated ARDS are potential candidates for angiotensin II.20,26,27,49
Clinical Discussion: Applying Angiotensin II in Practice
Emily See
See described vasodilatory shock as one of the most common, fatal conditions that clinicians manage in the ICU. Norepinephrine is the primary vasopressor for the treatment of vasodilatory shock in most patients;3 however, some patients are refractory to catecholamines, resulting in high-dose exposure and risk of toxicity.36 Angiotensin II can be used as part of a multi-modal vasopressor strategy to enhance efficacy and minimise toxicity risk.3
See stated that patients with hyperreninemia, prior exposure to ACE inhibitors, and severe AKI requiring RRT might derive the greatest benefit from angiotensin II treatment, as described above.20,24,25,26
Focusing on the practical aspects of angiotensin II use in clinical practice, See described that angiotensin II has a plasma half-life of less than 1 minute,52 making it readily titratable. It is diluted in saline to a final concentration of 5,000 or 10,000 ng/mL, and the solution can be stored either at room temperature or under refrigeration for up to 24 hours.52 Angiotensin II is administered through a central venous line at a starting infusion rate of 20 ng/kg/min.52 The dose range is 1.25–40 ng/kg/min (up to 80 ng/kg/min in the first 3 hours).52 Patients should be continuously monitored for blood pressure response.52
Angiotensin-II Initial MAP Response Index of Treatment Effect (AIMRITE) was developed as a tool to identify angiotensin II responders. It is calculated as the change in MAP at 1 hour from baseline, divided by the study drug dose.23 Patients with an AIMRITE of ≥0.9 mmHg/ng/kg/min are considered responders.23 Secondary analysis of the ATHOS-3 study showed that 60% of patients receiving angiotensin II had AIMRITE ≥0.9 mmHg/ng/kg/min (Figure 3A), and these responders had a lower risk of death compared with resistant patients who received angiotensin II (AIMRITE: <0.9 mmHg/ng/kg/min; p=0.0279), and those receiving placebo (p=0.0438; Figure 3B).23
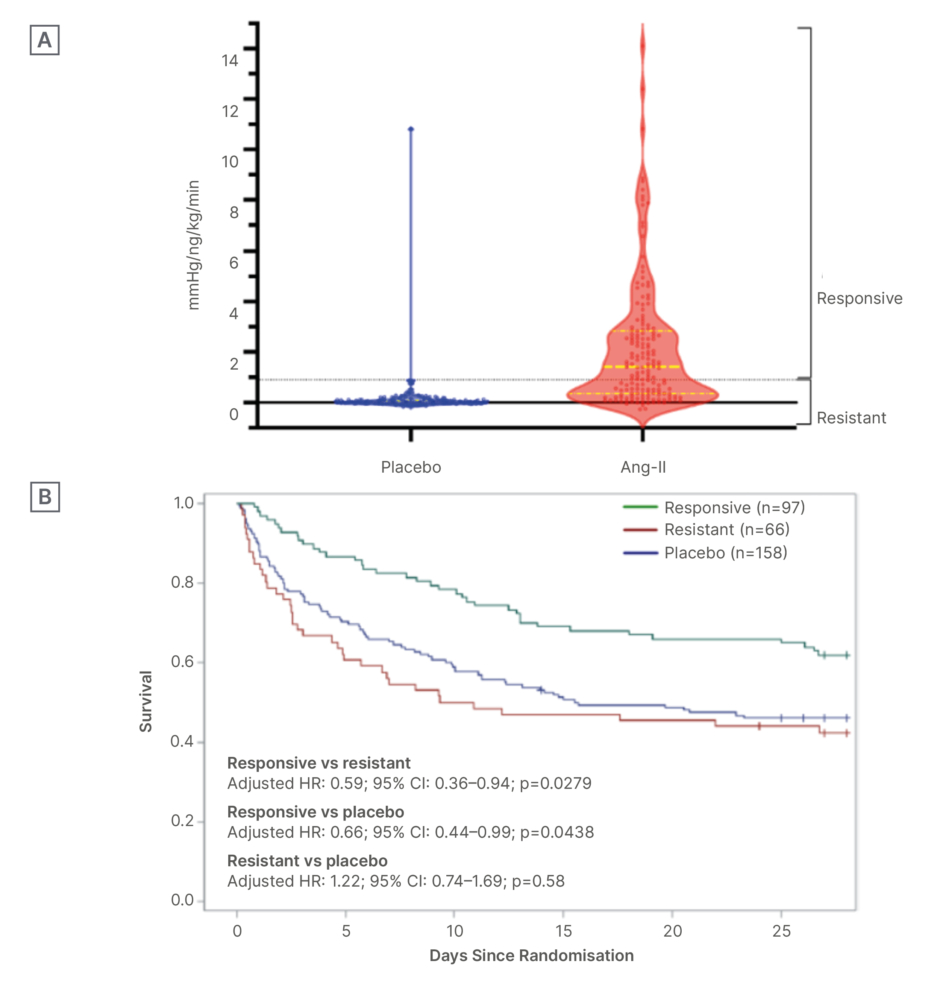
Figure 3: Angiotensin-II initial mean arterial pressure response index of treatment effect at 1 hour and survival according to treatment and response or resistance in the ATHOS-3 study.23
A) Distributions of initial MAP response by treatment assignment and AIMRITE phenotype: violin plots of initial MAP response at 1 hour, indexed to study-drug dose, stratified by treatment assignment. Dotted lines indicate thresholds used for assignment in categorical analyses. Dots correspond to individual patients. Highlighted solid and broken lines indicate the median and interquartile range, respectively.
B) Association of survival to Day 28 with initial MAP response to angiotensin II treatment: survival plots for angiotensin II responsive (green) versus resistant (red) versus placebo (blue) patients. Adjusted HRs show the indicated comparison from the primary analysis multi-variable model.
HR: hazard ratio; MAP: mean arterial pressure; vs: versus.
As vasodilatory shock improves, it becomes necessary to de-escalate vasopressors.
Initial safety concerns about thromboembolic events with angiotensin II were raised following the ATHOS-3 study.13 A systematic review of RCTs and observational studies that reported thromboembolic event rates revealed no difference in the pooled event rates between angiotensin II and placebo; however, high risk of bias precluded quantitative analysis, and the certainty of evidence was low.53 Despite these findings, the Giapreza Summary of Product Characteristics (SmPC) retains a warning for thromboembolic events and recommends appropriate venous thromboembolism prophylaxis during treatment.54
See summarised that applying angiotensin II in practice requires clinicians to identify eligible patients, consider special populations, start treatment at a modest dose and titrate accordingly, use a multi-modal vasopressor weaning strategy for down-titration, and keep abreast of emerging data on angiotensin II. Finally, See noted that there are several ongoing studies in Europe, the USA, and Australia, stating that “these important studies will really enrich our knowledge of how best to use angiotensin II in niche populations” (data available on request).
GL-GIA-2025-00040 | April 2026