Abstract
Myofibrillar myopathy (MFM) is a rare inherited neuromuscular disorder characterised by progressive muscle weakness with potential cardiac and respiratory involvement. Mutations in the LDB3 gene disrupt Z-disk integrity, leading to myofibrillar disintegration and protein accumulation. Reports of LDB3-related MFM are limited, particularly in Southeast Asian populations.
The authors describe a Filipino mother–daughter pair with genetically confirmed LDB3-associated MFM due to a novel variant. The proband developed progressive proximal muscle weakness beginning in childhood, while her daughter presented with proximal lower extremity weakness in early adulthood. Neither patient exhibited ophthalmoplegia or bulbar involvement. Electromyography and nerve conduction studies demonstrated a myopathic pattern with irritative potentials. Muscle biopsy of the proband revealed features consistent with MFM with multicore formation. Genetic testing identified a heterozygous LDB3 c.1804T>C variant in both individuals.
Supportive and rehabilitative management was initiated with ongoing monitoring for systemic complications. This report represents the first documented association of the LDB3 c.1804T>C variant with MFM, expands the phenotypic spectrum of LDB3-related disease, and highlights the importance of integrated clinical, pathological, and genetic evaluation in familial proximal myopathy, particularly in underrepresented populations.
Key Points
1. This report broadens the global understanding of myofibrillar myopathy (MFM) by describing the first documented Filipino case associated with a novel LDB3 variant (c.1804T>C, p.Tyr602His). It emphasises the relevance of identifying rare genetic variants and recognising diverse phenotypic presentations in underrepresented populations.2. The article presents a mother–daughter pair with MFM linked to the novel LDB3 variant, detailing longitudinal clinical features, electrophysiology, muscle biopsy findings, and genetic confirmation. The case illustrates both inter- and intra-familial variability in age of onset, severity, and progression of skeletal muscle involvement.
3. Clinicians should maintain early suspicion for MFM in patients with progressive proximal muscle weakness, particularly with a positive family history. Comprehensive evaluation, including genetic testing, is critical. Awareness of variable phenotypes within families supports accurate diagnosis, timely monitoring for potential cardiac involvement, and appropriate genetic counselling.
INTRODUCTION
Myofibrillar myopathies (MFM) are a heterogeneous group of inherited muscular dystrophies defined by a characteristic histopathological pattern rather than a single genetic aetiology. They are characterised by myofibrillar dissolution originating at the Z-disk, with subsequent sarcomere disintegration and accumulation of myofibrillar degradation products, often accompanied by ectopic protein expression and, in some cases, congophilic material. Clinically, MFMs exhibit marked phenotypic variability, most commonly presenting with progressive muscle weakness that frequently involves proximal muscle groups, with possible ophthalmoplegia, bulbar involvement, cardiomyopathy, and peripheral neuropathy.1-4
Mutations in LDB3, which encodes the Z-disk protein Z-band alternatively spliced PDZ-motif (ZASP), have been implicated in MFM and are associated with considerable clinical heterogeneity.1-4 However, LDB3-related MFMs remain infrequently reported, particularly in Southeast Asian populations. The authors describe a Filipino mother–daughter pair with genetically confirmed MFM due to a novel LDB3 c.1804T>C variant, expanding the genotypic spectrum of LDB3-associated disease.
CASE DESCRIPTION
A 45-year-old Filipino woman (proband) presented with progressive proximal muscle weakness, initially affecting the lower extremities and later involving the upper extremities. Her symptoms began in childhood, where she frequently stumbled and required more time to rise after falls compared with her peers, without associated myalgia, cramps, or numbness. During adolescence, she experienced easy fatigability during squatting and difficulty with prolonged running. In early adulthood, she developed progressive difficulty climbing stairs and occasional knee buckling, requiring assistance for high-stepping activities such as boarding public transport. She maintained independence in basic and instrumental activities of daily living until her early 30s, when worsening weakness and frequent falls necessitated assistance with ambulation and transfers. Weakness later progressed to the upper extremities, resulting in difficulty lifting her arms overhead, sitting up from a supine position, and standing from a chair. Family history was notable for a daughter who developed bilateral proximal lower extremity weakness at 21 years of age, initially presenting with difficulty lifting her thighs during ambulation and frequent stumbling (Table 1). There were no other affected family members. Psychosocial history was unremarkable.
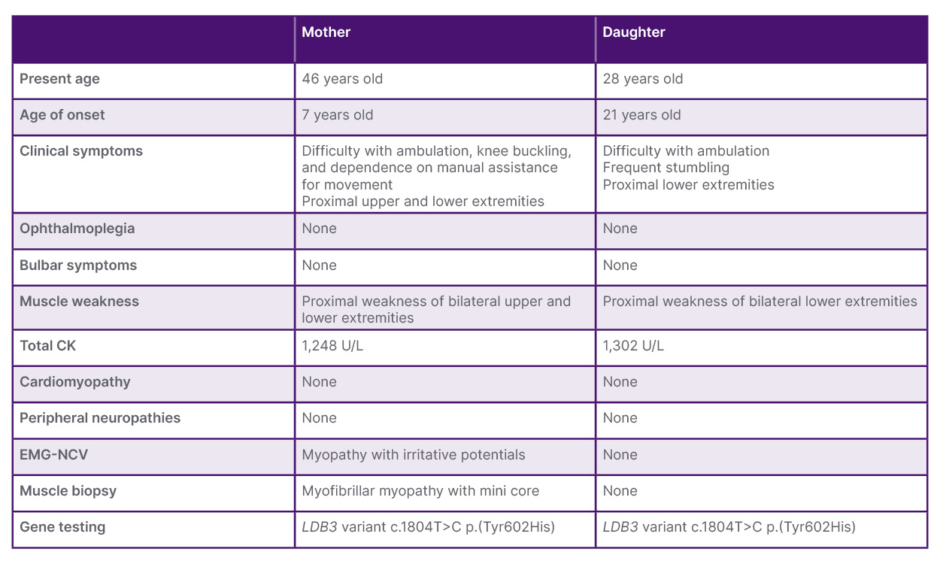
Table 1: Clinical profile of the proband and her daughter.
CK: creatine kinase; EMG-NCV: electromyography-nerve conduction velocity.
The neurologic examination revealed that the patient was alert and cognitively intact, with normal cranial nerves. Motor testing revealed symmetric proximal weakness in both upper and lower extremities, graded at 2/5, with relative distal sparing, and no fasciculations or myotonia were observed. Sensory examination was intact in all modalities. Cerebellar testing showed no dysmetria or dysdiadochokinesia. Deep tendon reflexes were 2+, and pathologic reflexes were absent.
Diagnostic workup revealed elevated serum creatine kinase levels. A 2D echocardiogram showed no evidence of cardiomyopathy. Electromyography and nerve conduction studies were consistent with a myopathic process with irritative potentials. Muscle biopsy of the biceps brachii demonstrated features of MFM with multicore formation (Figure 1 and Figure 2).
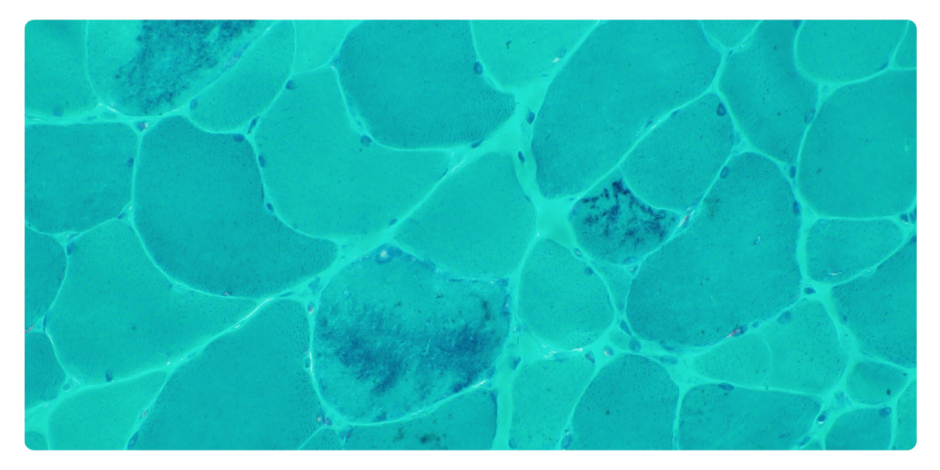
Figure 1: Modified Gomori trichrome with nemaline bodies.
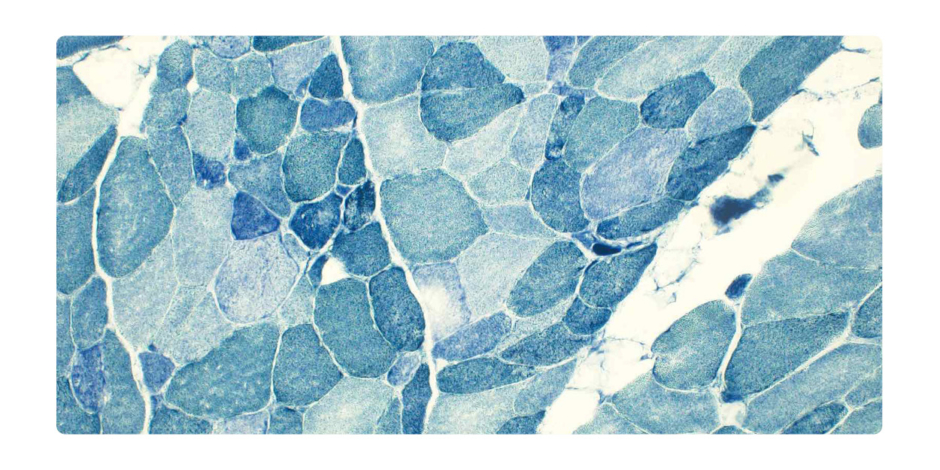
Figure 2: NADH showing disorganised intermyofibrillar networks and multi-mini cores.
NADH: nicotinamide adenine dinucleotide.
Genetic analysis was performed using next-generation sequencing (CentoXome® Solo, CENTOGENE, Rostock, Germany), which utilises exome capture and sequencing on an Illumina platform (Illumina, Inc., San Diego, California, USA) with >98% coverage of targeted regions. The identified LDB3 variant (NM_001171610.1:c.1804T>C; p.Tyr602His) was confirmed by Sanger sequencing. In silico prediction tools, including PolyPhen-2 and MutationTaster, suggested a deleterious effect, with high conservation across species. The variant is rare in population databases (Genome Aggregation Database [gnomAD] allele frequency: approximately 0.000008) and was identified in both affected family members, supporting segregation with disease.
The diagnosis of LDB3-associated MFM was established based on the combination of clinical, electrophysiological, histopathological, and genetic findings. Differential diagnoses, including other hereditary myopathies and limb-girdle muscular dystrophies, were excluded based on genetic testing and biopsy results.
Therapeutic interventions included pharmacologic and supportive strategies. The patient was prescribed co-enzyme Q10, one tablet twice daily, which she adhered to consistently. Supportive measures included mobility aids, supervised rehabilitation, and fall prevention strategies. No changes in therapy were made during follow-up, as co-enzyme Q10 was well-tolerated. Outcomes included stabilisation of proximal weakness without further short-term functional decline, though the patient remained dependent for transfers and ambulation. No adverse or unanticipated events occurred related to therapy. The prognosis is currently focusing on maintaining mobility, preventing complications, and monitoring systemic involvement, while the patient is being followed longitudinally to track disease progression and functional status.
DISCUSSION
MFMs are a genetically heterogeneous group of disorders characterised by Z-disc disorganisation and progressive myofibrillar degeneration, resulting in skeletal muscle weakness.1-4 Genes implicated in MFMs include DES, CRYAB, MYOT, FLNC, and LDB3, each associated with variable clinical phenotypes.1,2,5,6 The LDB3 gene encodes the ZASP protein, which plays a critical role in maintaining Z-disc structural integrity. Disruption of this protein leads to sarcomere instability, protein aggregation, and progressive muscle fibre degeneration.5,7,8
The identified LDB3 c.1804T>C (p.Tyr602His) variant is located within the C-terminal LIM3 domain, a functionally important region involved in protein–protein interactions, including binding to protein kinase C alpha (PKCα). Alterations in this domain may impair Z-disc integrity and contribute to myofibrillar disintegration and protein accumulation, supporting the biological plausibility of its role in disease pathogenesis.5,7,8
According to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines, the variant meets criteria, including PM2 (low population frequency), PP1 (co-segregation with disease), PP3 (multiple computational predictions supporting deleterious effect), and PP4 (phenotype highly specific for LDB3-associated MFM). Despite these findings, the variant remains classified as a variant of uncertain significance, highlighting the need for further functional studies to establish pathogenicity.
Clinically, the proband demonstrated early-onset proximal weakness beginning in childhood, while her daughter presented in early adulthood, illustrating both inter- and intra-familial phenotypic variability. This variability has been reported in other LDB3-related myopathies, which demonstrate a wide range of onset ages, severity, and organ involvement.5,6,9 Muscle biopsy findings, including fibre size variation, structural disorganisation, and core-like lesions, further support the diagnosis of MFM.1-4
LDB3 mutations have also been associated with cardiomyopathy, including hypertrophic cardiomyopathy, which may predispose patients to malignant arrhythmias and sudden cardiac death. Although cardiac involvement has been reported in a subset of patients with LDB3-related disease,5,7,8 both patients in this report underwent cardiac evaluation. Clinical examination did not reveal any cardiac abnormalities, with no audible murmurs on auscultation. Additionally, 2D echocardiography demonstrated no evidence of cardiomyopathy or structural heart disease. Nevertheless, given the recognised cardiac associations of LDB3 variants, continued longitudinal cardiac monitoring is recommended.
This report is strengthened by detailed clinical characterisation, histopathological confirmation, and genetic analysis in two affected first-degree relatives, supporting genotype–phenotype correlation. However, several limitations should be acknowledged. Functional studies were not performed to directly assess the impact of the variant on protein expression or ZASP isoform regulation. Additionally, extended familial genetic testing and long-term cardiac follow-up were not available.
Overall, this case highlights the importance of integrating clinical, pathological, and genetic findings in the diagnosis of MFM and contributes to the expanding spectrum of LDB3-associated disease.
CONCLUSION
This report expands the spectrum of LDB3-related myopathy by describing a novel LDB3 c.1804T>C variant with marked inter-and intra-familial phenotypic variability, highlighting the value of comprehensive clinical, pathologic, and genetic evaluation for accurate diagnosis and management.






