INTRODUCTION
Myocardial infarction with non-obstructed coronary arteries (MINOCA) is clinically defined by the evidence of myocardial infarction and macroscopical/visual appearance of normal coronary arteries at coronary angiography (any coronary artery stenosis more or equal to 50% in diameter), after excluding alternative diagnosis for troponin elevation such as Takotsubo syndrome, pulmonary embolism, sepsis, and myocarditis.1
MINOCA is a relevant yet commonly overlooked clinical problem for cardiologists, with a prevalence that can be as high as 10% among patients admitted for acute myocardial infarction and undergoing coronary angiography.1,2 Compared to those with myocardial infarction due to obstructive coronary artery disease (CAD), patients with MINOCA are often younger, especially female, and less frequently have a history of traditional cardiovascular risk factors.2
Of note, even if it may be considered relatively benign, MINOCA has a significant impact on 12-month mortality and risk of rehospitalisation, comparable to myocardial infarction due to obstructive CAD.3 Moreover, up to 25% of patients with MINOCA may experience recurrent angina episodes in the following 12 months.4 Recurrent angina can have a significant impact on healthcare-related costs, leading to repeated hospitalisations and invasive procedures, as well on patients’ quality of life, due to a higher probability of disability and premature retirement from the work.5 Therefore, an appropriate management of patients with MINOCA is of mainstay importance to improve patients’ prognosis, and to prevent negative socioeconomic consequences.
DISCUSSION
MINOCA represents an underinvestigated field of research, with some important unmet clinical needs due to the lack of an appropriate diagnostic and therapeutic consensus, and of specific pharmacological treatment. Some clinical trials are currently ongoing to answer these questions (Figure 1).
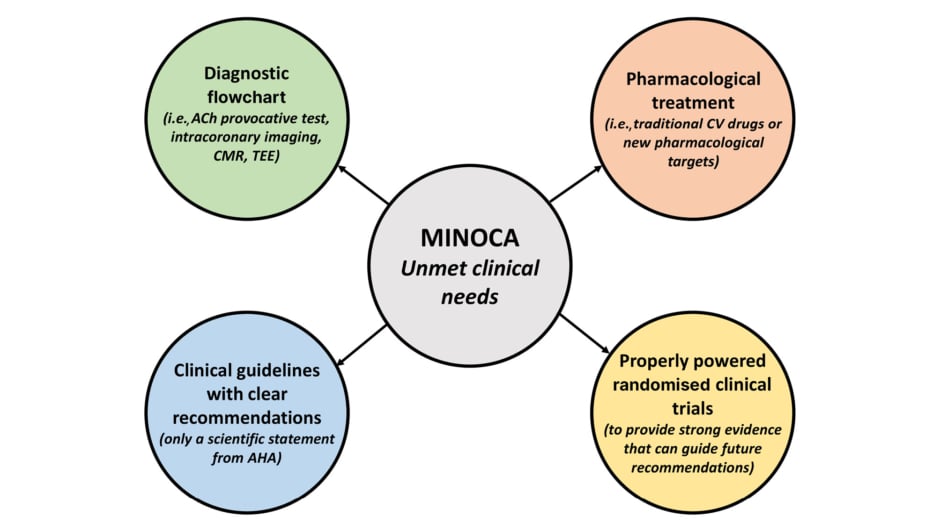
Figure 1: Schematic representation of the unmet clinical needs in myocardial infarction with non-obstructed coronary arteries.
ACh: acetylcholine; AHA: American Heart Association; CMR: cardiac magnetic resonance; CV: cardiovascular; MINOCA: myocardial infarction with non-obstructed coronary arteries; TEE: transoesophageal echocardiography.
Regarding the diagnosis, there is a wide range of potential pathogenetic mechanisms underlying MINOCA, including coronary plaque rupture (PR)/plaque erosion (PE) not determining angiographically flow-limiting stenosis (due to a transient thrombosis with spontaneous thrombolysis, distal embolisation, superimposed vasospasm, or a combination of these processes), spontaneous coronary artery dissection, epicardial and microvascular vasospasm, and coronary thromboembolism leading to microvascular obstruction.6 A pathogenetic characterisation is fundamental for the choice of the best medical approach in MINOCA: if treated uniformly as a ‘unicum’, each treatment does not have a uniform effect on the patient’s prognosis. For example, what may be beneficial for a specific subgroup of patients (i.e., dual antiplatelet therapy [DAPT] and β-blockers improving the prognosis in patients with PR or PE), may be useless or even counterproductive in the others (i.e., DAPT leading to an unbalanced increased risk of bleeding and β-blockers favouring coronary vasoconstriction by unmasking α-adrenoreceptors in patients with epicardial coronary spasm). Therefore, the term MINOCA should not be used to refer to a specific diagnosis, but instead, to a heterogeneous ‘working diagnosis’ in which the application of an appropriate diagnostic workup (including invasive and non-invasive tests) can progressively help to elucidate the underlying mechanism and implement the correct therapy (Figure 2).
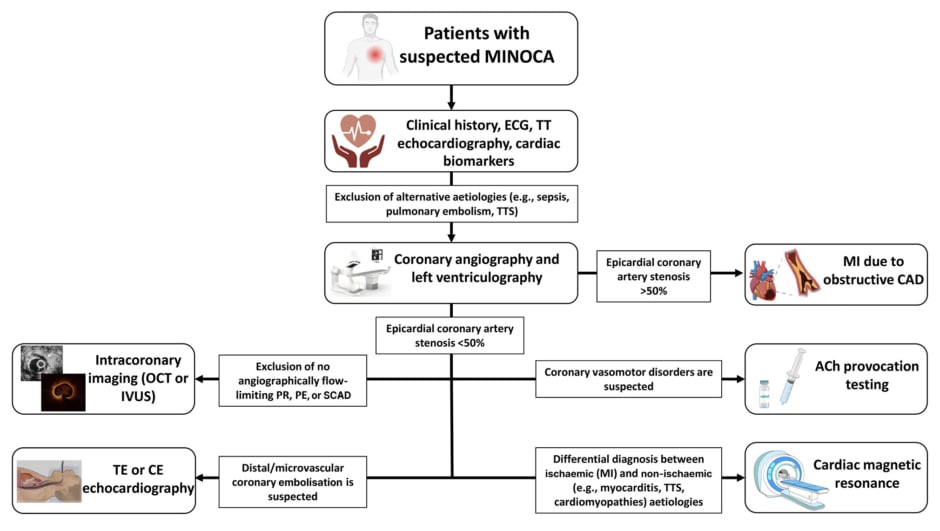
Figure 2: Diagnostic flowchart in patients with suspected myocardial infarction with non-obstructed coronary arteries.
ACh: acetylcholine; CAD: coronary artery disease; CE: contrast-enhanced; ECG: electrocardiogram; IVUS: intravascular ultrasound; MI: myocardial infarction; MINOCA: myocardial infarction with non-obstructed coronary arteries; OCT: optical coherence tomography; PE: plaque erosion; PR: plaque rupture; SCAD: spontaneous coronary artery dissection; TE: transoesophageal; TT: trans-thoracic; TTS: Takotsubo syndrome.
Several advanced diagnostic techniques beyond coronary angiography and transthoracic echocardiography should be considered in MINOCA. Intracoronary provocation testing with acetylcholine is fundamental for the diagnosis of functional coronary alterations (i.e., epicardial or microvascular spasm). It has been recently demonstrated that performing an acetylcholine provocation test in patients with MINOCA is safe and has important prognostic implications, as patients with a positive response are at increased risk of future cardiovascular events compared to those with a negative one.7 The use of intracoronary imaging, such as optical coherence tomography or intravascular ultrasound, can help to detect frequently unrecognised causes at coronary angiography (i.e., PR, PE, or spontaneous coronary artery dissection).8,9 Cardiac magnetic resonance, thanks to its high accuracy in discriminating between ischaemic or non-ischaemic aetiologies, may help in the differential diagnosis between myocardial infarction, inflammatory cardiac diseases (e.g., acute pericarditis or myocarditis), and Takotsubo syndrome. Transoesophageal echocardiography or contrast enhanced echocardiography may be helpful if distal/microvascular coronary embolisation is suspected based on the presence of risk factors, such as atrial fibrillation, mechanical valves, or thrombophilic disorders.
It is of note that many hospitals do not have such diagnostic workup well organised in their clinical practice yet, mainly because of lack of knowledge, or because there are no clear recommendations from clinical guidelines. Therefore, there is a need for an appropriate and validated diagnostic flowchart that should be implemented in clinical practice when managing patients with MINOCA, to avoid leaving the diagnosis to cardiologists’ discretion and/or expertise.10 To date, only the American Heart Association (AHA) has tried to summarise and unify the diagnostic alghorithm and therapy in MINOCA in a scientific statement. In this document, a ‘traffic light’ sequence for the diagnosis of MINOCA was proposed, but no clear indications about use of adjunctive tests to coronary angiography were given, likely because of limited evidence-based literature.2 Conversely, the most recent European Society of Cardiology (ESC) clinical guidelines for patients presenting with acute coronary syndromes do not specifically address and delineate appropriate management of MINOCA.11,12 Management of MINOCA should be properly addressed in the upcoming clinical guidelines or in appropriate scientific consensus documents, especially by the European societies, given the lack of European documents.
Furthermore, although identification of the underlying MINOCA aetiology may guide a proper and customised acute and long-term treatment, there are few data about what is the best pharmacological treatment. In a large study including 9,136 patients with MINOCA enrolled in the SWEDEHEART registry, the use of statins and renin-angiotensin system inhibitors led to a significant reduction in the rate of major adverse cardiovascular events (defined as all-cause mortality, hospitalisation for myocardial infarction, ischaemic stroke, and heart failure) at a mean follow-up of 4.1 years. A trend for a reduction of events was observed with the use of β-blockers, while DAPT, the cornerstone therapy for atherosclerotic obstructive CAD, had a neutral effect on clinical outcomes. However, the study cohort was extremely heterogeneous, as the specific pathogenetic mechanism leading to MINOCA was not identified and medical therapy was not modulated accordingly.13 Moreover, available studies mainly focused on evaluating the role of traditional CV drugs (i.e., angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, β-blockers, statins, and DAPT) in MINOCA, with only few ongoing RCTs investigating the best approach and pharmacological management. MINOCA-BAT is an ongoing clinical trial that aims to evaluate if a therapy with oral β-blockers or angiotensin-converting enzyme inhibitors/angiotensin receptor blockers may reduce the incidence of all-cause death, readmission because of myocardial infarction, ischaemic stroke, or heart failure in patients discharged after MINOCA with left ventricular ejection fraction ≥40%.14,15
StratMed-MINOCA16 is an ongoing clinical trial that will determine whether an early risk stratification by coronary microvascular dysfunction (defined by an index of microvascular resistance ≥25) associated with cardio-protective mineralocorticoid antagonist therapy with eplerenone could reduce the changes of levels of N-terminal pro-brain natriuretic peptide as marker of myocardial damage in patients with MINOCA. The ongoing PROMISE17 clinical trial will evaluate whether a ‘precision medicine approach’, defined as a comprehensive diagnostic workup associated with a consequent tailored pharmacological treatment for the underlying aetiology, compared to ‘standard of care’, consisting of coronary angiography alone and the standard treatment of myocardial infarction (DAPT in all patients, β-blockers, statins, and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers if clinically indicated), may improve patients with MINOCA’s prognosis and quality-of-life.18
Further studies are strongly warranted either to gain a deeper insight into the pathophysiology of MINOCA, or to explore new potential pharmacological targets in MINOCA such as circulating biomarkers (e.g., endothelin-1 and neuropeptide Y) involved in coronary vasomotion,19 platelet-derived soluble CD40-ligand and myeloperoxidase involved in plaque destabilisation,20 and small circulating non-coding RNA (e.g., microRNA involved in many cellular pathways, such as proliferation, angiogenesis, differentiation, and apoptosis).21
CONCLUSION
In conclusion, although the prevalence of MINOCA is likely to increase in the next years, the diagnostic and therapeutic approach of patients with MINOCA is still unclear and frequently left at cardiologists’ discretion, or based on the experience of each centre. Therefore, MINOCA represents an urgent unmet clinical need that should be properly addressed in future clinical guidelines or expert consensus documents.