
Abstract
Schnitzler syndrome is a rare acquired autoinflammatory disorder that is characterised by recurrent fevers, bone or joint pains, urticarial rash, and monoclonal immunoglobulin M paraprotein, while the variant form has immunoglobulin G monoclonal paraprotein. The cytokine that appears to cause the inflammatory episodes is interleukin-1β, and blocking this cytokine ameliorates almost all symptoms of this disorder. Physicians should be aware of this disorder so that they can recognise this difficult form of urticarial vasculitis and prevent the complication of amyloidosis.
INTRODUCTION
Schnitzler syndrome was first described by the dermatologist Dr Liliane Schnitzler in 1972.1 It is now recognised as a rare acquired autoinflammatory disease of unknown cause that presents with non-specific clinical signs and biochemical features of inflammation over many years (fevers, bone pains, urticaria rash, and classical immunoglobulin [Ig]M monoclonal paraprotein).2 The histopathological findings of skin biopsies reveal perivascular neutrophil and lymphocytic infiltration into the upper dermis and occasional diffuse neutrophilic infiltration in the middle dermis (neutrophilic urticaria). It is similar to lupus erythematosus, adult-onset Still’s disease, and, rarely, autoinflammatory syndromes of the cryopyrinopathies.
We describe a case of variant Schnitzler syndrome (with IgG monoclonal paraprotein), where the patient was treated initially for multiple myeloma, but the treatment was stopped as the urticarial rash progressed into a form of urticarial vasculitis, therefore raising the possibility of Schnitzler syndrome.
CASE DESCRIPTION
A 37-year-old female was admitted in the Hematology Department of the Apollo Gleneagles Hospital Kolkata, Kolkata, India, with history of recurrent low-grade fevers, flitting arthralgia, and blurring of vision for 1 month. Her medical history revealed generalised arthralgias with occasional multiple large joint pains for 3 years and anaemia for 2 years with recurrent low-grade fevers, for which she received multiple courses of antibiotics with little or no effect. Investigations elsewhere into the anaemia (haemoglobin 6.0 gm/dL) and raised erythrocyte sedimentation rate at 115 mm/first hour had revealed a small paraprotein (quantitated at 8.6 g/L) and 8% bone marrow plasma cells. Immunofixation showed IgG-lambda, for which she had received one dose of bortezomib and dexamethasone.
There was no history of Type 2 diabetes mellitus or hypertension. She was a non-smoker and denied alcohol or illicit drug use. There was no family history of periodic fevers, monoclonal gammopathy, or any family members admitted with pyrexia of unknown origin at any time.
Her laboratory results showed low haemoglobin at 6.1 g/dL, a white cell count of 9,500/mm3, platelet count of 216×109/L with elevated erythrocyte sedimentation rate at 48 mm/1st hour (Table 1). The findings of low serum iron and vitamin B12 levels confirmed the diagnosis of a mixed haematinic deficiency anaemia. She was started on broad-spectrum antibiotics that were discontinued after admission when infections were ruled out. Complete blood count after 3 days of admission showed white cell count 10,600/mm3 with neutrophilic pleocytosis (82%). The patient continued to complain of joint pains, but anti-cyclic citrullinated peptide antibody analysis was negative with normal X-ray of shoulder and knee joints.
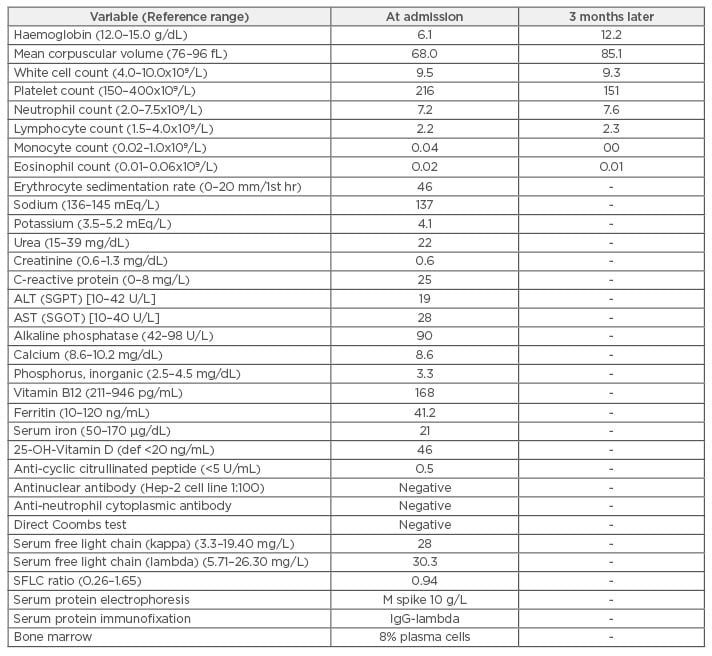
Table 1: Laboratory investigation results at admission and subsequent review.
ALT (SGPT): alanine aminotransferase (serum glutamic-pyruvic transaminase); AST (SGOT): aspartate
aminotransferase (serum glutamic oxaloacetic transaminase); def: deficiency; IgG: immunoglobulin G;
SFLC: serum-free light chain.
A rheumatological consultation led to the suggestion of a seronegative spondyloarthropathy and ophthalmological consultation indicated possible episcleritis. Following this, it was advised to start administering the patient Homide eye drops (homatropine-hydrobromide) twice daily and prednisolone acetate ophthalmic suspension eye drops.
A whole body bone scan (20 mCi of technetium 99m-methyl diphosphonate [99mTc-MDP] given intravenously and images obtained after 3 hours) showed increased tracer uptake in both sacroiliac joints, right more than left (Figure 1). Serum protein electrophoresis showed monoclonal gammopathy and serum protein immunofixation confirmed the paraprotein as IgG lambda. Positron emission tomography with computed tomography (PET-CT) imaging showed diffuse [18F]-2-fluoro-2-deoxy- D-glucose (FDG) uptake in both tonsils (standard uptake values [SUV] maxright 6.0 and left 5.6), mild FDG avid subcentimeter size bilateral cervical level IIA nodes were seen, the largest measuring 11×8 mm (SUV max 2.7), and mildly FDG avid left inguinal lymph node (SUV max 4.1), all of which were considered to be either infective/inflammatory in nature. FDG avid cysts were seen in both ovaries; the largest left ovarian cystic lesion measured 12×9 mm (SUV max 5.1) and was considered to be corpus luteal cysts. There was no abnormal uptake to suggest active metastatic disease or any myelomatous deposits.
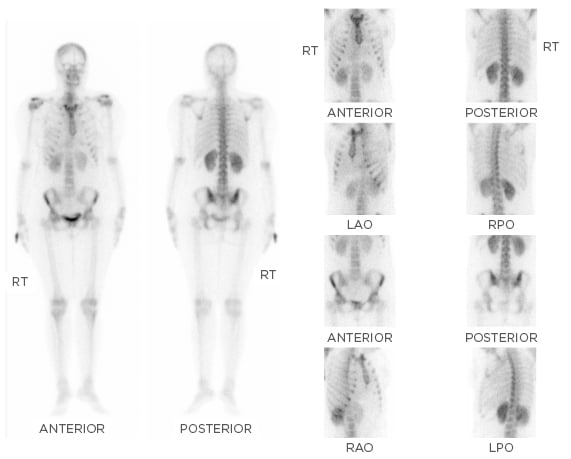
Figure 1: Whole body bone scan using 20 mCi of 99mTc-MDP with images obtained after 3 hours showing increased tracer uptake in both the sacroiliac joints, right more than the left.
99mTc-MDP: technetium 99m-methyl diphosphonate; LAO: left anterior oblique; LPO: left posterior oblique; RAO: right anterior oblique; RPO: right posterior oblique; RT: right.
As the patient was suffering from iron deficiency and vitamin B12 deficiency, she was given iron infusion and a single dose of vitamin B12 1 mg injection. She was discharged on folic acid 5 mg and ferrous gluconate tablets with a plan to review in the outpatient clinic.
She was seen 5 weeks after being discharged, when she complained of increased joint pains, recurrent diffuse pruritic maculopapular lesions over her face, abdomen, and upper extremities, with daily attacks over 4 weeks. She, however, had an excellent response to haematinic therapy with a rise in haemoglobin of 5 g/L. At this point, an immunology referral was made with a provisional diagnosis of urticarial vasculitis. The patient revealed that she now suffered outbreaks with variable intensity almost on a daily basis, with no identifiable triggering factors. She mentioned that the lesions cleared up in 24 hours, leaving no marks or scars, while new lesions had appeared almost daily. The physical examination at this time revealed diffuse pruritic maculopapular lesions over her face, thorax, and upper extremities, sparing her palms, soles, and mucous membranes. There was evident synovitis affecting both wrists, ankle, and knee joints, but no overt arthritis. She refused skin biopsy.
She was started on high-dose antihistamines and a 10-day course of oral steroids that controlled the urticaria. However, after 3 weeks, bone pains with fevers were reported almost on a daily basis (maximum temperature recorded was 400C) and urticaria relapsed with more of a burning than an itching sensation, which now lasted 48–72 hours (Figure 2).
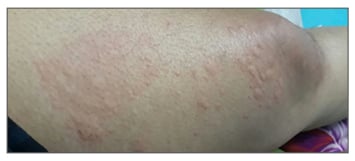
Figure 2: Urticarial rashes of variable nature that is characteristic of Schnitzler syndrome, as exhibited by our patient.
Based on the clinical presentations and laboratory results, the diagnosis of Schnitzler syndrome was made as she fulfilled the diagnostic criteria: both major criteria (chronic urticarial rash/intermittent fever and monoclonal paraprotein) and three out of five minor criteria (arthralgias, bone pain, and elevated erythrocyte sedimentation rate). There was no evidence of hepatosplenomegaly or lymphadenopathy and there was perhaps a transient phase of neutrophilic leucocytosis. She was counselled regarding the diagnosis of Schnitzler syndrome and discussed starting antiinterleukin 1 therapy (anakinra). Serum amyloid A and C-reactive protein levels were measured to ascertain disease activity at 10 weeks from diagnosis, which showed borderline raised serum amyloid A at 6.91 mg/L (normal, <6.4 mg/L) and normal C-reactive protein at <0.50 mg/L.
Anakinra could not be started due to financial constraints; colchicine 0.5 mg twice daily was started with long-acting non-sedating anti-H2 blocker fexofenadine 180 mg, which led to almost complete resolution of symptoms in about 6 weeks from starting treatment. She has since presented on two further occasions with florid urticaria requiring short-term steroids and remains on long-term colchicine therapy.
DISCUSSION
Schnitzler syndrome is categorised under orphan disease and remains largely underdiagnosed. Studies from Mayo Clinic, Rochester, Minnesota, USA, suggest that Schnitzler syndrome may be present in ≤1.5% of patients with a monoclonal IgM in serum,3 while the variant form with monoclonal IgG probably remains undiagnosed. The most comprehensive review to-date is by de Koning,4 who reviewed 281 cases of Schnitzler syndrome from 25 countries that showed a male:female ratio of 1.5, median age at onset of 51 years, with the delay in diagnosis exceeding 5 years in most cases. In a previous case series of 94 patients by the same author, only 5 patients (5.3%) experienced symptoms before the age of 35 years,5 while a recent paper of 11 patients from Belgium (between 1995 and 2015), reviewed using the Strasbourg criteria, had only 2 patients who presented before the age of 35 years and all had IgM paraprotein (kappa light chain in 10 patients, while only one had lambda light chain).6 Our case is probably the first from India and has other interesting features: a young age onset of Schnitzler syndrome (37 years compared to median age 51 years), episcleritis as an unusual presentation, variant form of IgG-lambda monoclonal paraprotein, and fever and monoclonal paraprotein preceding onset of urticaria. Our patient had opted for naturopathy as a treatment option for almost a year and was avoiding several foods in her diet, which would explain the relatively severe anaemia and excellent response to haematinic replacement therapy (i.e. nutritional anaemia), and objectively no evidence of bone marrow infiltration. It is, therefore, not unusual that the presence of the severe anaemia and monoclonal paraprotein had prompted physicians to initially use bortezomib and dexamethasone to treat as multiple myeloma, with the presence of anaemia and bone pains satisfying two of four ‘CRAB’ features (hypercalcaemia, renal impairment, anaemia, bone pains). As patients with Schnitzler syndrome may occasionally receive full courses of chemotherapy for multiple myeloma before the diagnosis of Schnitzler syndrome is suspected,7 awareness of this entity needs to be increased.
It is perhaps always a concern of physicians and patients whether chronic (idiopathic) urticaria may be a manifestation of an underlying malignancy, especially a haematological malignancy. Among known significant disease associations with urticaria, monoclonal gammopathy of undetermined significance (MGUS) appears to be associated with urticaria in about 144.8 per 100,000 patient-years.8 A study using the Mayo Clinic electronic database (1994–2001) of 1,639 patients presenting with chronic urticaria found 47 of 797 patients evaluated for existence of monoclonal protein had MGUS, 142 had a malignancy, and 24 had both.9 This study concluded that patients presenting with chronic urticaria at an older age (>56 years) were more likely to have associated underlying MGUS. The variant form of Schnitzler syndrome with IgG paraprotein has not been described extensively, apart from a few reports,10-14 but the clinical presentation and laboratory features of inflammation are very much similar to the well-described form with IgM paraprotein. A relatively new form of IgG monoclonal gammopathy-related acquired autoinflammatory syndrome (proposed as Mullins’ syndrome) is, however, distinct from Schnitzler syndrome with features of neutropenia, complement activation, and resistant to anti-IL1 (anakinra) therapy with no mutations in any of the known periodic fever-related syndrome genes.15
Almost all patients with Schnitzler syndrome will present with urticaria-like dermatoses at disease onset, which are typically recurrent, last 24–48 hours, and the symmetrical nature of the rash differentiates them from chronic spontaneous or the physical/inducible urticarias. These dermatoses present with more of a burning pain than itching and generally respond poorly to antihistamines. Our patient presented with urticarial lesions after the arthralgia, which has been documented in another report.6 The skin lesions did not show a temperature-related effect; hence, cryoglobulin testing was not performed. Angioedema is uncommon in this condition. Intermittent fever, not periodic, affects nearly 75% of patients and may range from daily episodes to a few times in a year. As there was no family history of periodic fevers, or fevers with features of amyloidosis, genetic testing for periodic fever related syndromes (such as Muckle-Wells syndrome or tumour necrosis factor-related autoinflammatory syndromes) was not done in this case. Joint pains usually precede the skin rash in Schnitzler syndrome, and involve larger peripheral joints (knees, hip, back) without arthritis. Bone pains affect >50% of patients and typically involve the shins, including long bones, the hips, and back. A bone scan is, therefore, a helpful screening tool if Schnitzler syndrome is suspected, with typical findings of osteosclerosis especially involving the knees and pelvis.16 There have been reports of patients with Schnitzler syndrome having unusual symptoms, such as intercostal neuralgia with headache and pancreatitis,4 which may be similar to the finding of episcleritis in our patient. We think that inflammation due to IL-1β can affect any site, as seen with patients with cryopyrin-associated periodic syndromes who develop episcleritis, and this manifestation may have been due to untreated Schnitzler syndrome. It is therefore important that complications of Schnitzler syndrome, as with any autoinflammatory syndromes/diseases, are avoided, for example, amyloid A amyloidosis. Furthermore, there should be mandatory follow-up to detect the development of haematological malignancies, such as Waldenström’s macroglobulinaemia.6
The treatment options of Schnitzler syndrome are outlined in Table 2, with most experts recommending that the first-line treatment for patients with significantly reduced quality of life or evidence of persistent elevation of inflammatory markers/high risk of amyloidosis should be anakinra or with canakinumab.17 Canakinumab is a human monoclonal anti-human IL-1β antibody of the IgG1/kappa isotype.18-20 It binds to human IL-1β and neutralises its activity by blocking its interaction with IL-1 receptors. We used colchicine as primary treatment option in our patient given the cost restraints with anakinra. Colchicine is a tricyclic alkaloid that inhibits microtubules and is the preferred treatment of choice in familial Mediterranean fever, where it markedly reduces episodes of fever and prevents amyloidosis; a similar mechanism of action may be at play in Schnitzler syndrome given the modest efficacy of this drug. Colchicine is also known to stabilise proteinuria in patients with amyloid nephropathy. However, it is now clear that symptoms in Schnitzler syndrome are most likely related to the monoclonal paraprotein and thus this disease should be classified under monoclonal gammopathy of cutaneous significance.21
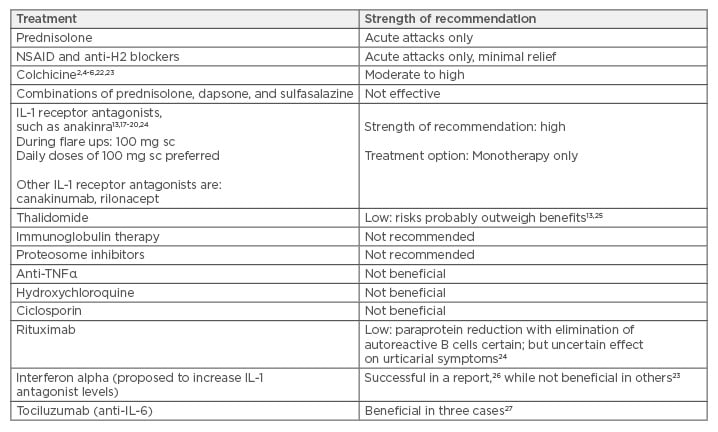
Table 2: Treatment options for Schnitzler syndrome with strength of recommendation.
Notes: Strength of recommendation is mainly based on case reports, case series, and single-centre
experience, as referenced.
IL: interleukin; NSAID: non-steroidal anti-inflammatory drugs; sc: subcutaneous; TNFα: tumour necrosis
factor alpha.
In our limited experience, we recommend colchicine as the first-line therapy for management of Schnitzler syndrome. Anti-H1 blockers and colchicine have now been prescribed long-term for our patient, with short rescue courses with corticosteroids when required, but continuous assessment with complete blood count, C-reactive protein, urine routine examination every 3 months, and paraprotein levels every 6 months has been recommended for long-term follow-up including monitoring for treatment-related complications.





