
Abstract
Haemangiomas are benign tumours growing by vascular endothelial hyperplasia, commonly occurring in females. The main types diagnosed in children are infantile haemangioma and congenital haemangioma, and these can also be found: in the liver, the gastrointestinal tract, intramuscularly, in vertebrae, intradurally, calvarially, and in the skull base. They can cause functional impairment, high output cardiac failure, and consumption coagulopathy, with current treatment options of corticosteroids, propranolol, embolisation, surgery, and laser treatment. Following a brief review of the literature, a rare case of a calvarial lesion is conveyed. A 57-year-old man presented with a right frontal parasagittal swelling and a computed tomography (CT) scan showed a lesion with a ‘honeycomb’ appearance. It was excised and the histopathological report received described the lesion as an intraosseous cavernous haemangioma.
INTRODUCTION
Haemangiomas are benign tumours which arise from the proliferation of endothelial cells surrounding blood-filled cavities. The female to male ratio observed is 3:1, with most haemangiomas presenting soon after birth with gradual involution. Often their rapid growth may interfere with the airways, gastrointestinal tract, and musculoskeletal function, or may cause high output cardiac failure and disseminated intravascular coagulopathy. Additionally, large lesions can trap platelets leading to thrombocytopenia (Kasabach–Merritt syndrome). Haemangiomas of the small bowel, lungs, liver, and mucous membranes can occur in inherited Osler–Weber–Rendu disease.
REVIEW OF THE LITERATURE
Vascular anomalies are congenital lesions of abnormal vascular development.1 Vascular anomalies were classified in 1982 by Mulliken and Glowacki2 into vascular tumours, which grow by cellular hyperplasia, and vascular malformations, which represent a localised defect in vascular morphogenesis. Haemangiomas are vascular tumours,1 with infantile haemangiomas being the most common tumour in infancy.
Infantile haemangiomas arise from haematopoietic progenitor cells in the appropriate milieu of genetic alterations and cytokines. Abnormal levels of matrix metalloproteinases (MMP) and proangiogenic factors (vascular endothelial growth factor [VEGF], basic fibroblastic growth factor [b-FGF], and transforming growth factor beta 1 [TGF-β1]) play a role in haemangioma pathogenesis. Infantile haemangiomas present shortly after birth as well-demarcated, flat, and erythematous red patches. They show three distinct developmental phases: proliferation, quiescence, and involution.
Earlier nomenclature referred to these manifestations as strawberry or capillary haemangioma, cavernous haemangioma, and capillary cavernous haemangioma. Haemangiomas can be superficial, deep, or compound: superficial haemangiomas are red and nodular with no subcutaneous component, a deep haemangioma presents as a protrusion with an overlying bluish tint or telangiectasia, and compound haemangiomas have both deep and superficial components. Nearly 10% of children require intervention because of bleeding, ulceration, visual axis obstruction, airway obstruction, high-output cardiac failure, or risk of permanent disfigurement. Kasabach–Merritt syndrome, a potentially life-threatening coagulopathy characterised by enlarging haemangioma with severe thrombocytopenia, is associated with kaposiform haemangioendothelioma, tufted angiomas, and rarely with congenital haemangiomas.3
Corticosteroids, interferon, and vincristine have been used successfully for substantial and life-threatening disease. Topical corticosteroids have minimal efficacy; intralesional triamcinolone (3 mg/kg) can decrease tumour size in 75% of cases and intralesional steroid treatment is an option for focal haemangiomas of the parotid, nasal tip, subglottis, and eyelid. Oral prednisolone 3 mg/kg/day for 1 month is useful for haemangiomas >4 cm. Surgical management involves excision, laser treatment, or both. For large lesions causing high output cardiac failure, embolisation can be considered for initial control of cardiac overload.
In 2008, regression of a facial haemangioma was noted in a child being treated with propranolol for obstructive hypertrophic myocardiopathy. Since then, propranolol has been introduced as a primary treatment for complicated haemangioma.4 Propranolol is effective during the proliferative phase of growth and a dosing regimen of 1 mg/kg/day is commenced initially, increased to 2 mg/kg/day at 1 week if this is well tolerated. The indications for therapy are ocular involvement, airway compromise, presence of ulceration, functional impairment, or a combination of the above. Well-documented side effects reported include hypoglycaemia, gastrointestinal upset, and bronchospasm.
Congenital haemangiomas arise in the fetus, are fully grown at birth, and do not have postnatal growth. The major types observed (depending on clinical progression) are: rapidly involuting congenital haemangioma, which involutes within the first few months of life, and non-involuting congenital haemangioma, which is persistent. Partially involuting congenital haemangiomas are congenital haemangiomas with overlapping features,5 and usually, these do not require resection, however this may be needed in some non-involuting congenital haemangiomas.
Liver haemangiomas are the most common benign liver tumours. These lesions are usually incidental findings of imaging studies of the abdomen performed for other reasons.6 Hepatic haemangiomas can be classified into three morphological patterns: focal, multifocal, and diffuse. Focal hepatic haemangiomas are the hepatic form of cutaneous, rapidly involuting congenital haemangiomas and focal haemangiomas are fully grown at birth, but regress faster during development. In contrast, multifocal and diffuse hepatic haemangiomas are true infantile haemangiomas, undergoing parallel phases of growth and involution.7 Liver haemangiomas can be readily diagnosed by ultrasound or multiphase contrast-enhanced helical computed tomography (CT) scans. The indications for surgical resection are progressive abdominal pain in combination with size >5 cm. Haemangioma of the upper gastrointestinal tract is a rare cause of bleeding.8 Bleeding is often recurrent and diagnosis is often delayed for months to years. It should be considered in patients with recurrent gastrointestinal bleeding and negative conventional investigations. Mesenteric angiography may be the only investigation which will identify these lesions and results after excision are excellent.
Chronic pain and a new mass are the most common presenting symptoms;9 intramuscular haemangiomas present with muscular pain in particular. Pain is often exacerbated with exercise of the involved muscle due to the vascular dilation and increased regional blood flow. Clinical findings that support the diagnosis of haemangioma include isolated pulsations, extremity enlargement when dependent and regression when elevated, compressibility, increased temperature, muscle contracture, tenderness on palpation, and muscle weakness. Magnetic resonance imaging (MRI) is the diagnostic procedure of choice, as it reliably differentiates haemangiomas from malignant tumours without the need for a biopsy. A T1-weighted axial MRI through the haemangioma is very slightly hyperintense to muscle and shows multiple interlacing vascular channels of the lesion. Conservative management is the first-line of treatment for nearly all isolated intramuscular haemangiomas and rapid tumour growth, intractable pain, risk of local skin necrosis, thrombocytopenia, cosmetic or functional impairment, or suspicion of malignancy may necessitate surgical intervention.
Vertebral haemangiomas are recognised as one of the commonest benign tumours of the vertebral column.10 These predominantly affect the thoracic and lumbar vertebrae, with cervical involvement being rarer. These can become symptomatic and cause neurological deficit through any of the following four reported mechanisms: i) epidural extension; ii) expansion of the involved vertebra causing spinal canal stenosis; iii) spontaneous epidural haemorrhage; and iv) pathological burst fracture. The lesion most frequently occurs in the vertebral body and the radiological features of vertebral haemangiomas includes reduced bone density between much denser vertical trabeculae. This ‘jailhouse’ pattern was reportedly based on the lamellar arrangement of cancellous bone around space containing vessels. Surgical intervention is warranted in vertebral haemangiomas presenting with neurological complications. Decompression of the cord can be accomplished with a laminectomy, or corpectomy with instrumentation.
Capillary or cavernous haemangiomas have been rarely encountered in the spinal intradural space and in the cauda equina. Clinical features present as lower back pain, radicular pain, paresthesias, decreased sensation, motor weakness, a positive Lasegue’s sign, urinary retention, retrograde ejaculation, and impotence.11-13 MRI reveals an intradural, well-circumscribed, contrast-enhancing mass, with a surgical approach performed through laminectomy. The appearance of the tumour is pinkish-red and is found attached to one root. Histologically, there is proliferation of capillary-sized vessels lined by flattened endothelial cells, indicating capillary haemangioma.
Intraosseous haemangiomas are rarely seen in the calvarium.14-16 The parietal bone is the most commonly involved, followed by the frontal bone and less frequently by the occipital and temporal bones. A history of trauma seems to be related in some cases reported within the literature. Loss of vision and proptosis have appeared when the bone lesion involves the orbital, mandibular, or maxillary bones. Excessive bleeding can occur as a major complication at the time of surgery and after tooth extraction. Facial nerve paralysis or hearing loss can develop with temporal bone involvement of intraosseous haemangioma, with CT scans showing osteolytic lesions replacing normal bone tissue. The differential diagnoses for intraosseous haemangioma include fibrous dysplasia, osteoma, Langerhans cell histiocytosis, and multiple myeloma. Fibrous dysplasia exhibits a ‘ground glass appearance’; on the other hand, intraosseous haemangioma demonstrates a ‘honeycomb’ appearance, which is the scattering of radiodense spots within the radiolucent hollow. The treatment of choice for intraosseous haemangioma is en bloc resection with an adequate normal bone margin and the bony defect can be reconstructed by various methods. Other therapeutic methods include curettage, radiotherapy, and embolisation.
Skull-base haemangiomas have been reported to arise from the occipital condyle, causing neck pain and torticollis, from the foramen magnum and clivus causing basilar impression, and from the orbitosphenoid ridge and orbital roof causing progressive proptosis and visual loss.17 In MRI the lesion usually appears mottled and heterogeneous with both increased and decreased signal intensities on both T1 and T2-weighted images. Grossly, it appears as soft, rubbery, vascular tissue separated by thin bony trabeculae. Intraoperatively, the lesion appears as ‘moth-eaten’ bony interstices or white coral filled with vascular soft tissue. Microscopically, intraosseous cavernous haemangiomas are composed of thin-walled vascular channels lined by a single layer of flattened endothelial cells interspersed among bony trabeculae. The surgical approach becomes more difficult for extensive skull base haemangiomas. Successful excision requires the appropriate microsurgical skull base approach and technique. Radiation therapy remains a viable option for unresectable skull base lesions or residual tumours from subtotal resections.
CASE REPORT
A 57-year-old man presented with right parasagittal swelling which had increased in size. It was measured as 2×2 cm, hard in consistency, and non-mobile. A CT scan showed mixed density swelling in the right high frontal bone, involving the diploë but not eroding into the intracranial space. The appearance was honeycomb-like, with radiodense spots in radiolucency (Figure 1). Surgery was performed under local anaesthesia with a longitudinal incision made. Hard nodular swelling was seen within the skull (Figure 2). It was ‘nibbled’ and drilled until normal-looking bone was seen around the area. Bone wax was used for haemostasis and the wound healed. Histopathological examination showed bony trabeculae separated by widened marrow spaces containing scanty marrow elements. The marrow was replaced by a cavernous type of vascular spaces lined by plump endothelial cells and filled with blood, diagnostic of intraosseous cavernous haemangioma (Figure 3).
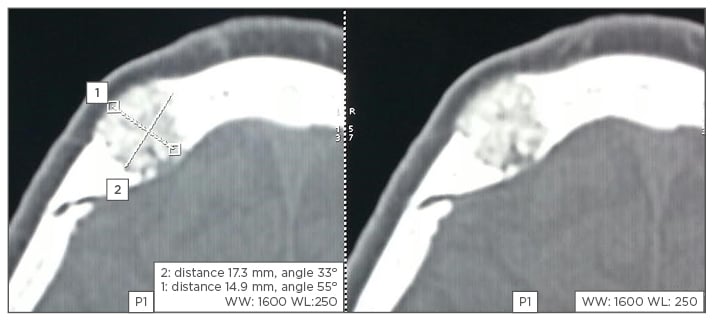
Figure 1: CT scan showing ‘honeycomb’ lesion of the skull.
CT: computed tomography.
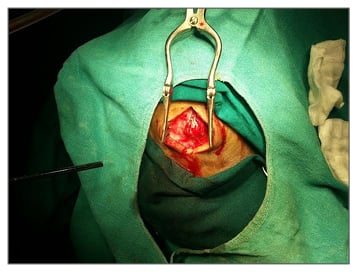
Figure 2: Preoperative picture of swelling.
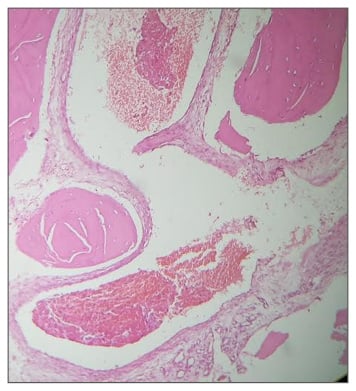
Figure 3: Histopathology of a cavernous haemangioma.
Histopathology performed by Dr Mary Mathew, Consultant Histopathologist, Muthoot Healthcare, Kerala, India.
CONCLUSION
Haemangiomas, although benign tumours of the vascular endothelium, can cause functional impairment and life-threatening complications. Medical, surgical, and embolisation treatments are current options. Haemangiomas of the liver, gastrointestinal tract, muscle, vertebra, cauda equina, calvarium, and the skull base have been described, although skull haemangiomas are rare; the treatment for these is excision.





