Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterised by loss of immune tolerance, widespread inflammation across multiple organs, and unpredictable flares, which lead to irreversible damage over time. Treat-to-target strategies and guideline-based care recommend treatment with hydroxychloroquine, glucocorticoids, immunosuppressants, and targeted biologics. However, despite these treatments, many patients continue to experience ongoing disease activity and treatment-related toxicities.
This article summarises key immune pathways driving SLE pathogenesis, including nucleic-acid sensing, Type I interferon (IFN) amplification, immune-complex/complement-mediated injury, and neutrophil extracellular trap (NET)-related inflammation. It also highlights immune trafficking and spatial organisation as determinants of organ involvement, focusing on chemokine networks and the sphingosine-1-phosphate–sphingosine-1-phosphate receptor (S1P–S1PR) axis as upstream regulators of immune localisation. Building on this rationale, S1PR1 modulation is discussed as an emerging therapeutic strategy, with studies on cenerimod progressing from proof-of-concept to Phase III trials. While multiple immune pathways are reviewed to reflect the complexity of SLE, particular emphasis is placed on immune trafficking and the S1P–S1PR1 axis as an upstream regulatory pathway under active clinical investigation. This review does not aim to provide an exhaustive catalogue of all emerging therapeutic modalities in SLE, but instead focuses on immune trafficking and spatial immune organisation as upstream regulators of disease.
ONGOING UNMET CLINICAL NEEDS IN SLE
SLE is a chronic autoimmune disease characterised by loss of immune tolerance, multisystem inflammation, and unpredictable flares, resulting in heterogenous organ involvement and long-term damage.1,2 SLE can involve multiple organ systems, including the vasculature, central nervous system, lungs, skin, kidneys, and joints, reflecting its underlying biological heterogeneity.3,4 Patients with SLE typically experience substantial impairment in quality of life, increased morbidity, and premature mortality.1,5 Despite advances in disease monitoring and supportive care, a substantial proportion of patients with SLE continue to experience ongoing disease activity, recurrent flares, and reduced survival.2
Over the last decade, efforts to optimise SLE management have increasingly focused on adopting a treat-to-target approach, in which disease remission is targeted through regular review and sequential treatment adjustment.6,7 Current guideline-informed management emphasises prompt initiation of treatment aimed at achieving remission or low disease activity in order to prevent flares and organ damage, improve prognosis, and enhance quality of life.2,7,8 In clinical practice, hydroxychloroquine is widely regarded as the cornerstone of therapy, with glucocorticoids used for rapid suppression of disease activity and tapered to the lowest feasible dose.2,8 Immunosuppressive agents, including mycophenolate mofetil, azathioprine, methotrexate, and biological agents are also commonly used to achieve and maintain control, particularly in patients with persistent moderate-to-severe disease (Figure 1).2,8
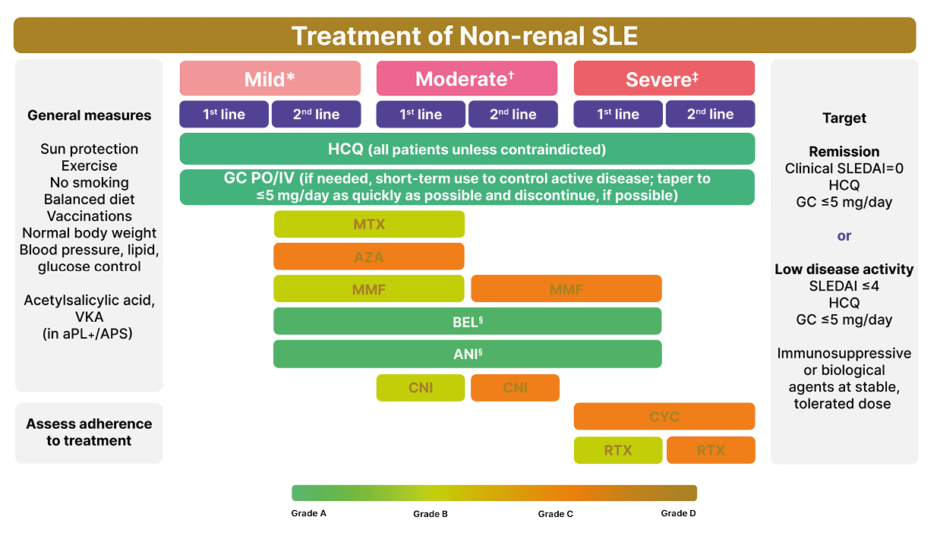
Figure 1: The European Alliance of Associations for Rheumatology (EULAR) recommendations for the treatment of non-renal SLE (2023 update).8
*Mild disease: constitutional symptoms; mild arthritis; rash ≤9% BSA; PLT: 50–100×109 /L; SLEDAI ≤6; BILAG C or ≤1 BILAG B manifestation.
✝Moderate disease: moderate-severe arthritis (‘RA-like’; rash: 9–18% BSA; PLTs: 20–50×109 /L; serositis; SLEDAI 7–12; ≥2 BILAG B manifestations).
‡Severe disease: major organ threatening disease (cerebritis, myelitis, pneumonitis, mesenteric vasculitis); thrombocytopenia with platelets <20×109 /L; TTP-like disease or acute haemophagocytic syndrome; rash >18% BSA; SLEDAI >12; ≥1 BILAG A manifestations.
§Recommendation of belimumab and anifrolumab as first-line therapy in severe disease refers to cases of extrarenal SLE with non-major organ involvement, but extensive disease from skin, joints, and so on. The use of anifrolumab as add-on therapy in severe disease refers mainly to severe skin disease. For patients with severe neuropsychiatric disease, anifrolumab and belimumab are not recommended.
Top-to-bottom sequence does not imply order of preference (e.g., MTX, AZA, and MMF are equal options for second-line therapy in mild disease or first-line therapy in moderate disease).
Adapted from Fanouriakis et al.8
ANI: anifrolumab; aPL: antiphospholipid antibodies; APS: antiphospholipid syndrome; AZA: azathioprine; BEL: belimumab; BILAG: British Isles Lupus Assessment Group; BSA: body surface area; CNI: calcineurin inhibitor; CYC: cyclophosphamide; GC: glucocorticoids; HCQ: hydroxychloroquine; IV: intravenous; MMF: mycophenolate mofetil; MTX: methotrexate; PLT: platelet count; PO: per os; RA: rheumatoid arthritis; RTX: rituximab; SLE: systemic lupus erythematosus; SLEDAI: SLE Disease Activity Index; TTP: thrombotic thrombocytopenic purpura; VKA: vitamin K antagonists.
Although steroids are effective for rapid symptom control in SLE, long-term reliance is associated with cumulative toxicity, infections, metabolic complications, cardiovascular morbidity, osteoporosis, and irreversible damage.9,10 Consequently, achieving effective steroid-sparing disease control without compromising safety remains a central priority for the management of SLE.4,8
Recently approved targeted therapies have expanded treatment options in SLE, particularly through modulation of B cell survival and Type I IFN-driven inflammation.4 However, persistent disease activity and the ongoing burden of treatment-related toxicity highlight the need to broaden therapeutic strategies beyond a limited number of dominant immune pathways.4 Increasing attention has therefore been focused towards upstream and under-recognised mechanisms, including immune cell trafficking and tissue-level regulation, as sources of future therapeutic innovation.11,12 These pathways offer a mechanistic rationale for emerging targeted approaches that intervene earlier in the inflammatory cascade, prior to amplification by downstream cytokine and effector pathways.11,13-15
KEY IMMUNE PATHWAYS IMPLICATED IN SLE
Systemic Immune Dysregulation as the Foundation of SLE Pathogenesis
SLE is characterised by chronic activation of innate and adaptive immune pathways.16 This chronic inflammatory state is shaped by defects in immune tolerance, persistent nucleic-acid sensing, and dysregulated cytokine networks that together reinforce autoreactivity and systemic immune imbalance.16-18 A central mechanism involves activation of nucleic-acid sensing pathways, including endosomal Toll-like receptors (TLR7 and TLR9), cytosolic RNA sensors such as retinoic acid-inducible gene I (RIG-I)-like receptors, and inflammasome pathways. These sensing processes promote ongoing production of Type I IFNs, cytokines that contribute to immune activation, antigen presentation, and downstream amplification of inflammation.16,18
In parallel, impaired clearance of apoptotic and necrotic debris increases exposure to nuclear antigens, facilitating immune-complex formation and propagation of autoantibody-driven inflammation.16,17 Complement deficiencies affecting early classical pathway components, particularly C1q, C2, and C4, are strongly associated with SLE because they impair immune-complex handling and clearance of apoptotic debris, further perpetuating inflammatory signalling and tissue injury.17 Together, these systemic abnormalities create a persistent pro-inflammatory environment in which autoreactive immune responses can be initiated and sustained.
Networks of DNA, histones, and neutrophil proteins, known as NETs, that are implicated in the capture of microbes may also be involved in the pathogenesis of SLE by promoting autoantibody formation and serving as adjuvants.16,17 Finally, environmental influences, including UV radiation, viral infection such as Epstein–Barr virus, and epigenetic dysregulation, may further increase susceptibility to immune activation.3,18
While these systemic abnormalities are necessary for disease initiation, they are insufficient to fully explain the organ specificity, persistence, and relapsing nature of tissue inflammation in SLE, and further pathological mechanisms must also be considered.
Immune Cell Trafficking and Spatial Organisation of Inflammation in SLE
Multisystem involvement in SLE depends not only on immune activation but also on the ability of leukocytes to migrate, localise, and persist within target tissues, where they contribute to chronic damage and functional impairment.13,19 Increasing evidence supports the concept that immune cell trafficking and spatial immune organisation are key determinants of disease expression and organ involvement.20 In this context, chemokine–chemokine receptor networks (e.g., CXCR3, CCR5, and their ligands) regulate tissue-specific immune cell recruitment and retention, contributing to organ-specific inflammation in SLE.20
Alongside chemokine-mediated recruitment, the S1P–S1PR axis represents a complementary regulatory system governing immune cell distribution.13-15,21-23 The S1P chemotactic gradient is a principal determinant of lymphocyte egress from secondary lymphoid organs into the lymphatic and vascular circulation via S1P–S1PR1 receptor engagement on B and T lymphocytes.23 In SLE, dysregulation of the S1P–S1PR axis has been proposed to alter immune cell distribution in ways that sustain both systemic and tissue-specific inflammatory activity, highlighting immune localisation as a key determinant of disease expression (Figure 2).14, 22,23
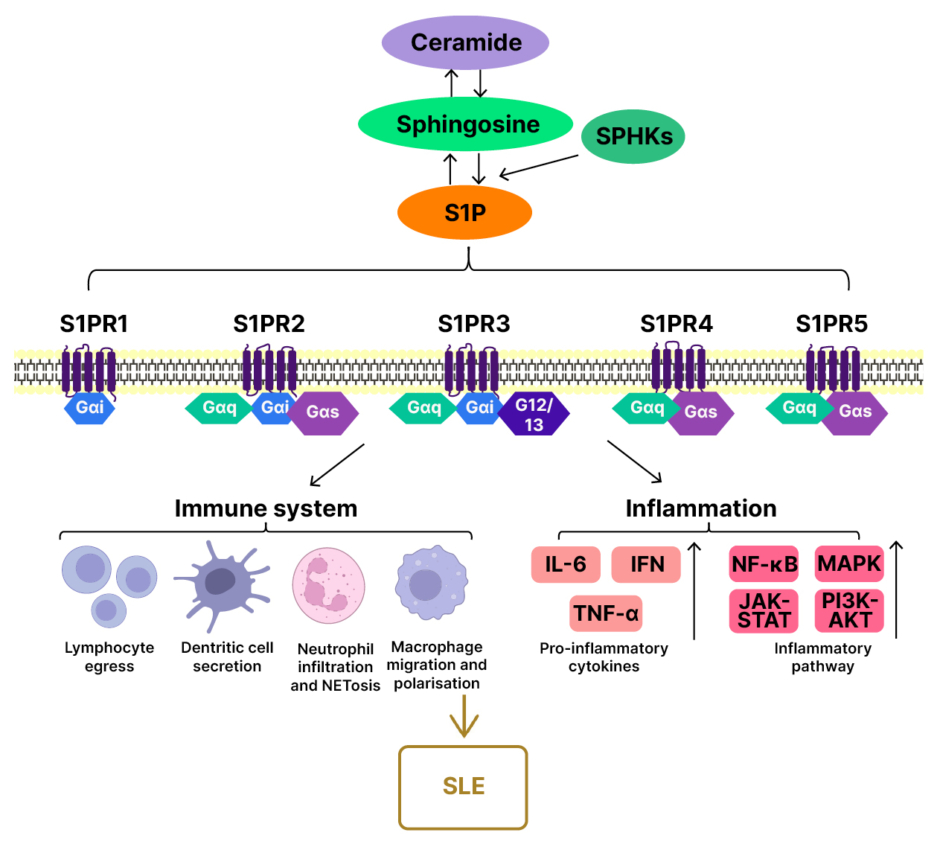
Figure 2: S1P signalling in the pathogenesis of SLE.14
In SLE, S1P functions as an important mediator by regulating both inflammatory and immune responses. On one hand, S1P plays a detrimental role by interacting with different immune cells. The spatial gradient of S1P drives lymphocytes into the peripheral circulation; thereby blockade of the S1P axis may mitigate the autoimmunity severity of SLE. Moreover, S1P is able to influence the function of innate immune cells, such as dendritic cells, neutrophils, and macrophages. On the other hand, as an inflammation-mediated disease, non-resolving inflammation has been regarded as the main contributor to SLE pathogenesis. Indeed, S1P can accelerate the inflammatory condition, which is likely associated with the crucial role of S1P in regulating pro-inflammatory cytokine secretion and signalling pathway activation.14
Adapted from Tian et al.14
G12/13: G protein alpha subunits 12 and 13; Gαi: inhibitory G protein alpha subunit; Gαq: Gq family G protein alpha subunit; Gαs: stimulatory G protein alpha subunit; IFN: interferon; JAK-STAT: Janus kinase-signal transducer and activator of transcription pathway; NETosis: neutrophil extracellular trap formation; NF-κB: nuclear factor kappa B; PI3K-AKT: phosphoinositide 3-kinase–protein kinase B pathway; S1P: sphingosine-1-phosphate; S1PR: sphingosine-1-phosphate receptor; SLE: systemic lupus erythematosus; SPHK: sphingosine kinase.
Among the five known S1PR subtypes (S1PR1–5), S1PR1 is highly expressed on naïve and central memory T cells, B cells, and subsets of dendritic cells, where it plays a dominant role in regulating lymphocyte egress from secondary lymphoid organs along S1P gradients. In contrast, S1PR2 and S1PR3 are more broadly expressed across immune and structural cell types, and may exert distinct or context-dependent effects, including influences on vascular tone and permeability.
S1PR signalling may also shape SLE pathology through effects that extend beyond lymphocyte trafficking. In addition to regulating immune cell distribution, S1PR activity in endothelial and other structural cells can influence tissue-level inflammatory responses by modulating vascular permeability, barrier integrity, and inflammatory cell extravasation.23
Endothelial S1PR1 Signalling and Tissue-Level Immune Regulation
Endothelial S1PR1 is emerging as a key regulator of vascular integrity and immune cell entry into inflamed tissues, with direct relevance to multisystem pathology in SLE.24,25 Through its actions on endothelial barrier stability, S1PR1 limits permeability and regulates leukocyte extravasation, thereby shaping the extent and persistence of tissue inflammation.26
Disruption of endothelial barrier function may contribute directly to organ-specific pathology in SLE by increasing vascular permeability and facilitating inflammatory cell extravasation into target tissues.26 Although S1PR signalling is increasingly recognised in vascular biology as a key pathway supporting endothelial integrity, its relevance to tissue-level inflammation in rheumatic disease remains under-appreciated.24-26 In parallel, dysregulated immune trafficking can increase the recruitment and retention of leukocytes at sites of injury.20 Together, aberrant immune localisation and impaired endothelial barrier control create a permissive environment for sustained immune cell-tissue interactions, reinforcing local cytokine signalling and antigen exposure.24,27 These conditions may then enable adaptive immune abnormalities to persist and evolve, driving chronic inflammation and long-term autoimmunity.
In this context, S1PR1 modulation may influence not only systemic lymphocyte redistribution but also tissue-level inflammatory dynamics. By stabilising endothelial barrier integrity and regulating leukocyte extravasation, S1PR1 signalling may limit the intensity and persistence of immune-cell infiltration within target organs.24,25 Although indirect, these effects provide a plausible mechanistic basis by which S1PR1-targeted therapies could attenuate local immune activation in addition to altering circulating lymphocyte numbers.
Endothelial dysfunction in specific SLE conditions, including lupus nephritis, neuropsychiatric SLE, and cutaneous disease, may reflect a shared driver of organ-specific inflammation, mediated through S1PR1 signalling. Framing S1PR1 as a molecular bridge between systemic immune dysregulation and tissue-specific pathology may therefore help align rheumatology perspectives with those of nephrology and neurology, and support therapeutic strategies aimed at restoring barrier function while simultaneously limiting pathological immune cell recruitment.
Dysregulated Lymphocyte Activation and Adaptive Immune Imbalance
Adaptive immune dysregulation is a hallmark of SLE and is central to autoantibody production, immune-complex formation, and organ injury.16 When B cell tolerance is disrupted, autoreactive B cells escape elimination and become activated, promoting autoantibody production and amplifying chronic inflammation.16,28 B cell activating factor (BAFF)/B lymphocyte stimulator (BlyS) overexpression supports survival and differentiation of autoreactive B cells and contributes to aberrant B cell activation and maturation.16 In addition, hyperactivation of nucleic-acid sensing pathways, including TLR7, can promote nucleic-acid-specific autoantibody production and contribute to plasmablast expansion, reinforcing humoral autoimmunity.16
T cell abnormalities contribute further to disease persistence through dysregulated cytokine production and impaired immune regulation. Expansion of Th17 cells and elevated IL-17 expression have been associated with disease activity and may contribute to neutrophil recruitment and increased inflammatory activity.16,17 Quantitative and functional deficiencies in regulatory T cells represent another pathway by which immune tolerance is impaired in SLE.17 Finally, production of most pathogenic IgG lupus autoantibodies occurs in the context of germinal centre reactions that require T follicular help cell-mediated support.11
Amplification Loops Sustaining Chronic Inflammation in SLE
SLE is characterised by amplification loops that perpetuate inflammation even when individual pathways are partially suppressed.29 Type I IFN signalling enhances antigen presentation, lymphocyte activation, and cytokine dysregulation, and may contribute to a self-reinforcing environment supporting chronic immune activation.29 Immune complexes and complement activation further reinforce tissue injury and promote recruitment of inflammatory cells, strengthening the persistence of organ inflammation.17,28 NET formation and IFN signalling may also interact to create self-sustaining inflammatory circuits that maintain autoantigen exposure and innate immune activation over time.16,17
These interconnected pathways may help explain why many patients experience persistent disease activity and the limited durability of response to therapies targeting downstream pathways.28 Collectively, therefore, these mechanistic insights underscore the rationale for therapeutic strategies that intervene upstream in immune organisation and trafficking. Specifically, the central role of the S1P–S1PR axis in regulating immune cell distribution provides a mechanistic foundation for clinical approaches targeting S1PR1 in SLE.14,15 Emerging translational and early-phase clinical data demonstrating reductions in IFN-associated biomarkers and autoantibody titres further support the biological plausibility of targeting this pathway.
CLINICAL DEVELOPMENT OF TARGETED THERAPIES IN SLE: FOCUS ON S1PR1 MODULATION
Overview of Targeted Therapeutic Strategies in Development
Investigational approaches in SLE are increasingly mechanism-driven, with therapeutic strategies targeting immune activation across multiple levels. These include IFN pathway inhibition, alongside B cell and T cell modulation through targeting B cells and key co-stimulatory pathways.3,9,16,27 Intracellular signalling inhibition is also represented, including kinase-directed approaches under advanced clinical evaluation.3,9,16,27 In addition, immune trafficking modulation is being explored via therapies targeting the S1P–S1PR1 axis to alter immune cell distribution and localisation.9
In this context, modulation of immune ‘spatial organisation’ has emerged as a conceptually distinct strategy.30 Instead of neutralising a single effector cytokine or depleting a specific immune population, trafficking-based approaches aim to alter the distribution of immune cells across lymphoid compartments, circulation, and peripheral tissues.30 This mechanistic strategy is supported by increasing evidence that immune cell localisation and persistence within target tissues influence organ involvement and flare dynamics in SLE.31-34
S1PR modulators are particularly notable because they target a pathway that regulates lymphocyte trafficking, thereby preventing them from migrating to sites of inflammation.35 Consequently, S1PR1 modulation provides a rational bridge between upstream immune organisation and downstream inflammatory outcomes in SLE.
Cenerimod: Mechanistic Rationale
Cenerimod is an oral S1PR modulator developed as a selective S1PR1 modulator for use in SLE.36 Its selectivity for S1PR1 (with activity at S1PR5) is intended to preferentially modulate lymphocyte trafficking while minimising S1PR3-associated cardiovascular effects observed with less selective S1PR modulators. In principle, S1PR1 modulation can reduce pathogenic lymphocyte recirculation by limiting lymphocyte egress from secondary lymphoid organs, thereby reducing the availability of circulating autoreactive lymphocytes that can enter tissues and sustain inflammatory responses.37 This mechanism offers wider immune modulation affecting many types of lymphocytes rather than the modulation of a single cytokine.
A key mechanistic advantage of this approach is that it alters immune cell distribution rather than directly eliminating immune cells. This is intended to reduce pathogenic trafficking while maintaining immune competence and surveillance.38 This is particularly relevant in SLE, where infection risk, cumulative immunosuppression, and treatment toxicity are critical considerations in long-term management.
Preclinical studies have shown that cenerimod causes dose-dependent changes in lymphocyte distribution and immune activity, consistent with its expected pharmacodynamic effects.39 In mouse models of autoimmunity, cenerimod improved survival and reduced circulating inflammatory lymphocytes. In murine lupus models, treatment reduced proteinuria and renal involvement, while in experimental autoimmune encephalomyelitis models, it reduced pathological changes in the brain.37,39
Clinical Trial Development of Cenerimod in SLE
Clinical development of cenerimod in SLE has progressed from proof-of-concept studies, through dose-finding evaluations, and into pivotal Phase III programmes23,36 (Table 1). In an early Phase II randomised trial (NCT02472795), cenerimod demonstrated dose-dependent reductions in lymphocyte counts over 12 weeks, consistent with its expected mechanism of action and pharmacodynamic effects.23 Exploratory clinical outcomes were also evaluated, supporting biological activity and informing subsequent development strategies.23
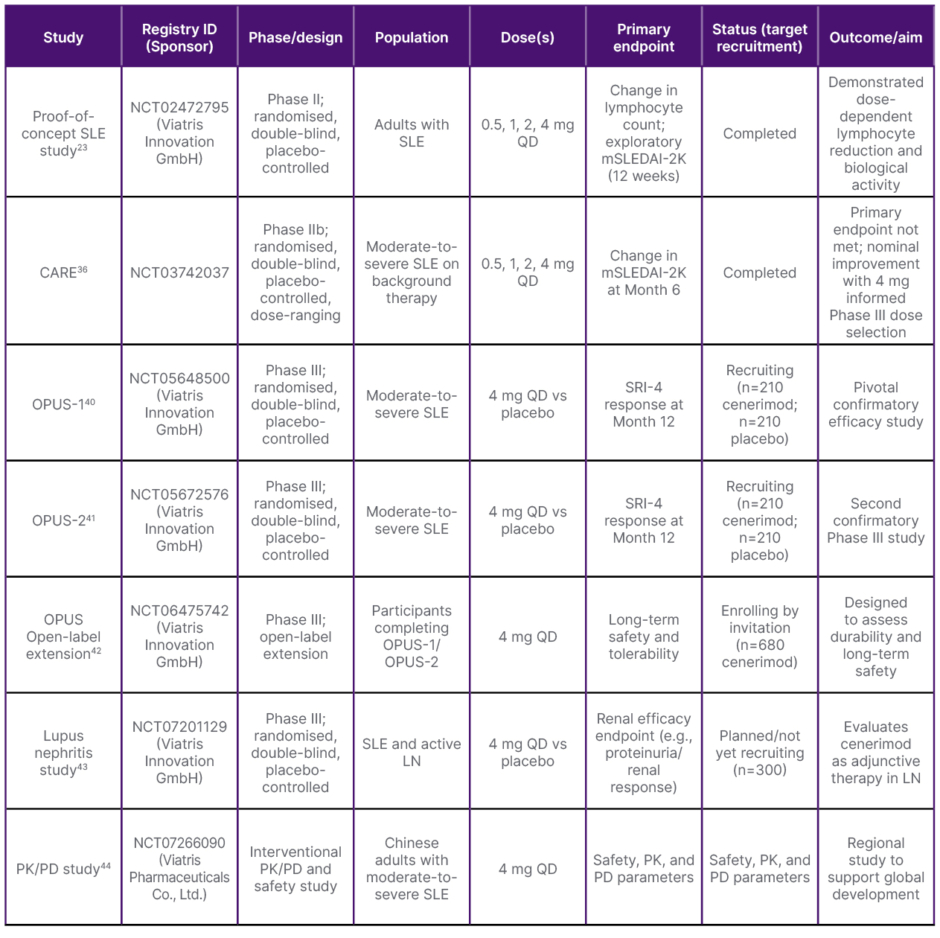
Table 1: Clinical studies of cenerimod in systemic lupus erythematosus.
LN: lupus nephritis; mSLEDAI-2K: modified Systemic Lupus Erythematosus Disease Activity Index 2000; PD: pharmacodynamics; PK: pharmacokinetics; QD: once daily; SLE: systemic lupus erythematosus: SRI-4: Systemic Lupus Erythematosus Responder Index 4; vs: versus.
Pharmacodynamic analyses from early-phase studies indicate that cenerimod reduces circulating cluster of differentiation (CD)4+ and CD8+ T lymphocytes as well as B lymphocytes, consistent with S1PR1-mediated sequestration of recirculating lymphocyte populations.23,35,45 Available data suggest broadly comparable reductions across T cell and B cell compartments rather than selective B cell depletion, reflecting its mechanism of action on lymphocyte egress rather than lineage-specific targeting.23,45 Naïve and central memory subsets, which are highly dependent on S1PR1 signalling for recirculation, appear particularly affected.15
The CARE Phase IIb trial (NCT03742037) evaluated cenerimod in patients with moderate-to-severe SLE receiving background therapy. In this randomised, double-blind study, the primary endpoint was the change in modified Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) at Month 6. Although the primary endpoint was not met, nominal improvement with the 4 mg dose informed selection of the dose used in Phase III trials.36,45
On the basis of these data, the ongoing Phase III studies, OPUS-1 (NCT05648500) and OPUS-2 (NCT05672576), are evaluating the efficacy and safety of cenerimod 4 mg once daily compared with placebo on top of standard of care, with Systemic Lupus Erythematosus Responder Index 4 (SRI-4) response at Month 12 as the primary endpoint.40,41 In addition, an OPUS open-label extension study (NCT06475742) is designed to assess long-term safety and tolerability, addressing the need for sustained safety evaluation in a chronic disease population with long-term treatment exposure.42 As the OPUS-1 and OPUS-2 trials are ongoing and blinded, comprehensive safety data, including detailed characterisation of adverse events, including infections, have not yet been publicly reported.
A key consideration for S1PR modulation in SLE relates to infection risk and background immunosuppression. Concerns around the safety of previous S1PR modulators have been raised in previous clinical trials in other autoimmune conditions, particularly with respect to infection risk.46,47 A meta-analysis of studies in multiple sclerosis reported that fingolimod was associated with a 16% increase in the risk of infection, especially lower respiratory infection and herpes virus infection.46 However, a study of fingolimod 0.5 mg/d for up to 5 years in patients with primary progressive multiple sclerosis showed that long-term infection risk was low, with 54 incidences of all infections over 100 patient-years.48 In terms of SLE, the CARE study showed that cenerimod was well tolerated by patients over a period of 12 months.36 Furthermore, although cardiac events have been linked to S1PR3 activity, because cenerimod is S1PR1/5-selective, cardiac risk may be expected to be lower than with less selective agents, supporting a more favourable cardiovascular safety profile.35,49,50 The results of the OPUS open-label extension study will provide further data to allow a full assessment of the long-term safety and tolerability of cenerimod in SLE.42
Many patients with SLE have baseline lymphopenia, raising an important safety consideration for therapies that further reduce circulating lymphocytes. As cenerimod lowers peripheral lymphocyte counts through S1PR1-mediated sequestration, individuals with pre-existing severe lymphopenia may be expected to require exclusion or additional monitoring to mitigate infection risk.
Finally, in preclinical SLE models, treatment reduced anti-double-stranded DNA (dsDNA) levels and lowered IFN-associated and pro-inflammatory biomarkers, including tissue readouts.23 In early clinical studies, cenerimod was associated with reductions in circulating anti-dsDNA titres and decreases in Type I IFN-related activity, including plasma IFN-α and an IFN-associated, whole-blood gene expression profile.23,35 Together, these findings suggest that S1PR1 modulation may attenuate both autoreactive humoral activity and IFN-driven pathways in SLE.
Although S1PR1 modulation primarily reduces lymphocyte egress from lymphoid tissues, the downstream immunological consequences are likely to extend beyond simple sequestration.15 By limiting recirculation of autoreactive T follicular helper cells and B cells, S1PR1 modulation may reduce germinal centre activity and plasmablast differentiation, thereby attenuating ongoing autoantibody production.11,32 In addition, reductions in circulating Type I IFN activity observed in early studies suggest that altered immune-cell trafficking may indirectly dampen IFN-driven B cell activation. The observed decreases in anti-dsDNA titres are therefore likely to be multifactorial, reflecting modulation of adaptive immune activation dynamics rather than redistribution alone.23,45
Positioning S1PR Modulation Within the Evolving SLE Treatment Landscape
S1PR modulation represents a mechanistically distinct approach compared with B cell targeted therapies or IFN pathway inhibition, as it targets immune cell localisation and recirculation upstream of many effector pathways.23,35 In principle, this strategy may allow broader immunomodulation across multiple immune cell subsets by reducing the probability of sustained immune activation within tissues and disrupting trafficking-dependent reinforcement of inflammation.
Potential advantages of this approach include upstream regulation of immune cell localisation and tissue-level inflammation in multiple cellular compartments, which may be relevant across diverse organ involvement where immune infiltration and endothelial dysfunction contribute to pathology.35,51 This may be particularly relevant for the long-term management priorities in SLE, including steroid-sparing disease control and reduction of chronic inflammatory burden.2,8-10
As with any emerging mechanism, the clinical value of S1PR modulation will depend on demonstration of meaningful clinical benefit, durability of response, and an acceptable long-term safety profile in the context of background immunosuppression. Ongoing Phase III trials will be critical in defining efficacy across clinical domains, identifying appropriate patient populations, and clarifying how S1PR modulation may be integrated into treatment algorithms in relation to other targeted therapies.
OVERALL PERSPECTIVE AND FUTURE DIRECTIONS
Current therapies for SLE have meaningfully expanded and now include guideline-supported use of immunomodulatory agents and targeted biologics.2,8 However, available approaches primarily address a limited subset of dominant immune pathways, leaving mechanisms such as immune trafficking, endothelial regulation, and innate-adaptive crosstalk incompletely targeted.3,15-18,20,22,24,25 This mechanistic gap aligns with persistent unmet needs in routine practice, including incomplete durability of response, ongoing flare burden, and sustained reliance on glucocorticoids in many patients.5
Mechanistic insights into SLE increasingly support a more integrated model in which immune activation is sustained not only by cytokine pathways, but also by the spatial organisation of immune responses. Within this evolving landscape, S1PR modulators expand available strategies by targeting immune cell organisation and localisation, rather than focusing only on the suppression of activation pathways. Cenerimod, a selective S1PR1 modulator, is an example of this approach in SLE and is currently being evaluated in Phase III trials.36,37,45 If ongoing studies confirm clinically meaningful efficacy, acceptable safety, and steroid-sparing potential, S1PR modulation could be an additional or sequential treatment option for patients who remain uncontrolled with existing therapies, especially when immune trafficking contributes to multisystem involvement.
Future progress in SLE therapeutics will depend on better translating disease mechanisms into effective clinical strategies. This includes integration of mechanistic and clinical trial data and developing a clearer evidence base for combination or sequential targeting of complementary pathways. In parallel, continued investigation into under-recognised mechanisms such as trafficking and tissue-level immune regulation may enable an earlier and more durable interruption of inflammatory cascades, ultimately improving long-term outcomes and reducing the overall treatment burden.