Abstract
Medullary carcinoma of the pancreas is an extremely rare malignant tumour. To the best of the authors’ knowledge, only 26 cases have been reported in the medical literature. A case of medullary carcinoma of the pancreas treated surgically by distal splenopancreatectomy is reported here.
Key Points
1. An extremely rare cancer, medullary carcinoma of the pancreas has, to the best of the authors’ knowledge, only been reported 26 times and has been classified by the World Health Organization (WHO) as a subtype of pancreatic ductal carcinoma.2. A literature review of the previous 26 cases indicated that the wild-type KRAS gene was found in 15 cases and six other cases had mutated KRAS, suggesting that this is an identifiable inherited cancer syndrome, where the identification of medullary carcinoma of the pancreas is essential.
3. The authors believe that, due to its rarity, an international database on treatment for patients with medullary carcinoma of the pancreas would be beneficial to patients.
INTRODUCTION
An extremely rare subtype of pancreatic adenocarcinoma that has recently been described is medullary carcinoma of the pancreas. The first reported case of this type of tumour was by Goggins et al.1 in 1998 and, since then, only 25 other cases have been reported in the medical literature. Medullary carcinoma of the pancreas is currently classified by the World Health Organization (WHO) as a subtype of pancreatic ductal carcinoma.1 However, medullary carcinoma differs from traditional pancreatic ductal carcinoma due to is its special genetic profile, as it has been reported that approximately 69% of medullary carcinoma tumours have wild-type KRAS genes and 22% have microsatellite instability (MSI).2 From a histological perspective, medullary carcinoma of the pancreas is described by the presence of highly pleomorphic cells with syncytial morphology, expansive tumour growth, and necrosis.1 Given its rarity, there are no universal guidelines for treatment and its management is extrapolated from the management of pancreatic ductal carcinoma. Here, a case of a 47-year-old male with medullary cancer in the body of their pancreas is presented.
CASE PRESENTATION
A 47-year-old male patient with no personal or family history of malignancy presented to the clinic for work-up of epigastric pain. On physical exam, a mild tenderness in the epigastric area was notable. Radiologic work-up with abdominal ultrasound showed enlarged lymph nodes on the coeliac trunk, and a CT scan of the abdomen and pelvis with intravenous contrast revealed a hypodense lesion in the body of the pancreas measuring 40×25 mm, with no evidence of vascular invasion (Figure 1). Magnetic resonance cholangiopancreatography showed a pancreatic body lesion measuring 30×25 mm, with heterogeneous density and delayed contrast uptake (Figure 2). An endoscopic ultrasound revealed a well-defined mass in the pancreatic body measuring 35×21 mm, which was hypoechoic, non-infiltrative, and inhomogeneous with central enhancement.
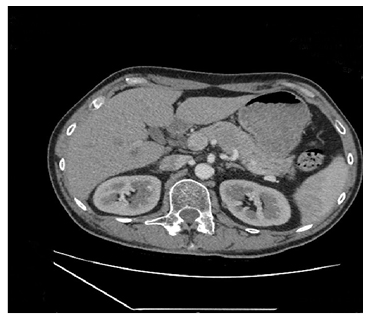
Figure 1: CT scan revealing a hypodense lesion in the of body of the patient’s pancreas measuring 40×25 mm.
The lesion had no evidence of vascular invasion and there was no dilation of the pancreatic duct.
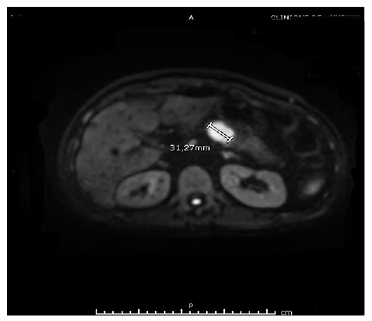
Figure 2: Magnetic resonance cholangiopancreatography showing a pancreatic body lesion measuring 30×25 mm, with heterogeneous density and delayed contrast uptake.
Multiple biopsies were taken, and histopathology revealed a non-invasive intraductal tubular papillary neoplasm. A PET-CT scan was performed, and no distant metastases were identified (Figures 3 and 4). Afterwards, a laparoscopic distal splenopancreatectomy was performed. Histopathology showed a 4.0×3.8 cm mass in the body of the pancreas that was compatible with medullary carcinoma, with negative surgical margins and perineural and perivascular invasion (Figure 5A). Microscopic examination showed a poorly differentiated carcinoma with limited gland formation, syncytial growth pattern, and stroma characterised by abundant tumour-infiltrating lymphocytes (Figures 5B and 5C). The borders of the tumour were well-defined in three-quarters of its periphery, where it was surrounded by a fibrinous pseudo-capsule (Figure 5D), and in the other quarter, there was an aspect of infiltrating stroma.
Figure 3: CT (left) and PET (right) scans of the patient’s abdomen showing a hypermetabolic lesion of pancreatic body (arrow) with no distant metastasis.
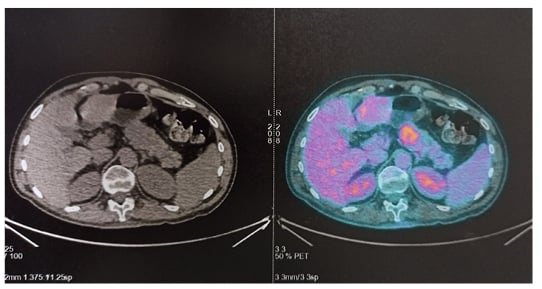
Figure 4: CT (left) and PET (right) scans of the patient’s pancreas showing a hypermetabolic lesion of the pancreatic body with no distant metastasis.
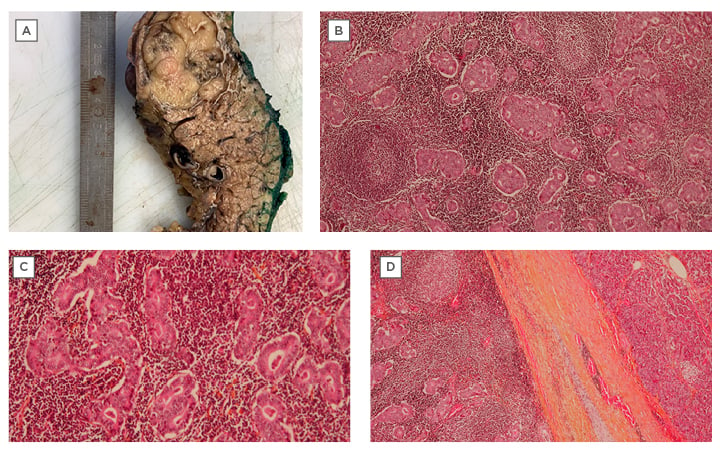
Figure 5: Histopathology performed after laparoscopic distal splenopancreatectomy.
A) Macroscopic image of the tumour after formalin fixation, measuring 4.0×3.8 cm. B) Microscopic image showing the syncytial growth and formation of glandular masses, which represent the tumour proliferation. The stroma has a basophilic appearance characterised by abundant lymphocyte proliferation forming lymphocyte follicles with a clear centre (arrows). C) Microscopic image showing the syncytial growth and formation of glandular masses, which represent the tumour proliferation. D) A fibrinous pseudo-capsule, covering most of the circumference of the tumour.
Immunohistochemistry for the neuroendocrine markers, cytokeratin-7, caudal-related homeobox transcription factor 2, B cell lymphoma 10, and cytokeratin-20 were negative. Tests for mutations of the EGFR (exons 18–21), KRAS (exons 2 and 34), NRAS (exons 2, 3, and 4), BRAF (exons 11 and 15), KIT (exons 8, 9, 11, 13, 14, 17, and 18), and PI3KCA (exons 10 and 21) genes were all negative. Henceforth, the tumour was considered to have a genetic profile of wild-type KRAS. No MSI was identified (phenotype: microsatellite stability [MSS]), which does not favour a diagnosis of Lynch syndrome. In situ hybridisation for Epstein–Barr virus peptide nucleic acid was also negative. Cytokeratin-19 was positive, and antigen Ki-67 proliferation was 30–40% in favour of an aggressive neoplasm. Moreover, an additional molecular study in search of a hypermutated tumour in the POLE gene profile was performed and came back as negative. After 12 weeks post-surgery, the patient started adjuvant chemotherapy with folinic acid plus fluorouracil plus irinotecan plus oxaliplatin and was disease-free up to 7 months post-surgery.
DISCUSSION
Medullary carcinoma of the pancreas is a recently described rare subtype of pancreatic adenocarcinoma with a special genetic profile.2 Its true incidence, prognosis, and optimal treatment are yet to be determined, and its management is currently extrapolated from the management of pancreatic ductal carcinoma. However, medullary carcinoma of the pancreas is genetically distinct from conventional ductal adenocarcinomas of the pancreas. In fact, Goggins et al.1 found that 60% of patients with pancreatic medullary carcinoma had both MSI and a wild-type KRAS gene. These data are atypical for non-medullary ductal adenocarcinomas of the pancreas, which nearly universally harbour KRAS gene mutations and seldom, if ever, have MSI.1,3 Its diagnosis depends on histologic findings, and there are no specific imaging or tumour markers that could aid these diagnoses. Consequently, pathologists should differentiate between medullary carcinoma and the conventional ductal adenocarcinoma as this will highlight the possibility of an inherited cancer syndrome, such as hereditary non-polyposis colorectal cancer, which will translate directly into better genetic counselling.
To the best of the authors’ knowledge, medullary carcinoma of the pancreas has been reported in 26 cases (Table 1). The mean age at diagnosis is 65 years, with 60% of patients being male and 40% being female, and 59% of these cases had a positive family history of cancer. However, regarding the anatomical location of the tumour, the case outlined here is the fourth reported case located in the body of the pancreas. Furthermore, despite the lack of information for all cases, it is worth mentioning that a personal history of colon cancer was in reported in three cases.
Medullary carcinoma of the pancreas suggests two distinctive genetics: MSI and a wild-type KRAS gene. In the reported cases, six had MSI, 18 had MSS, two had no MSI testing done, and one was of unknown status; therefore, MSI was found in almost 22.22% of cases (six out of 26 cases), compared with 66.66% who had MSS. As for the KRAS gene, the literature showed a preservation of wild-type KRAS in 15 cases, while six had mutant KRAS, four were of unknown status, and in one case, no KRAS analysis was performed. The majority of cases of medullary carcinoma of the pancreas were with a wild-type KRAS (15 out of 26; 55.55%), and a KRAS-mutated gene was seen in six out of 26 cases (22.22%). In current oncology practice, where cancer genetics and the correlation between the genotype and phenotype of the cancer play a vital role, the identification of an inherited cancer syndrome is of the utmost importance. From here, the role of the pathologist in identifying medullary carcinoma of the pancreas is essential and should be considered as the cornerstone for further investigations and genetic counselling. Furthermore, a high index of suspicion is necessary despite a negative diagnosis of malignancy on endoscopic ultrasound, so surgical consultation should be considered in these cases.
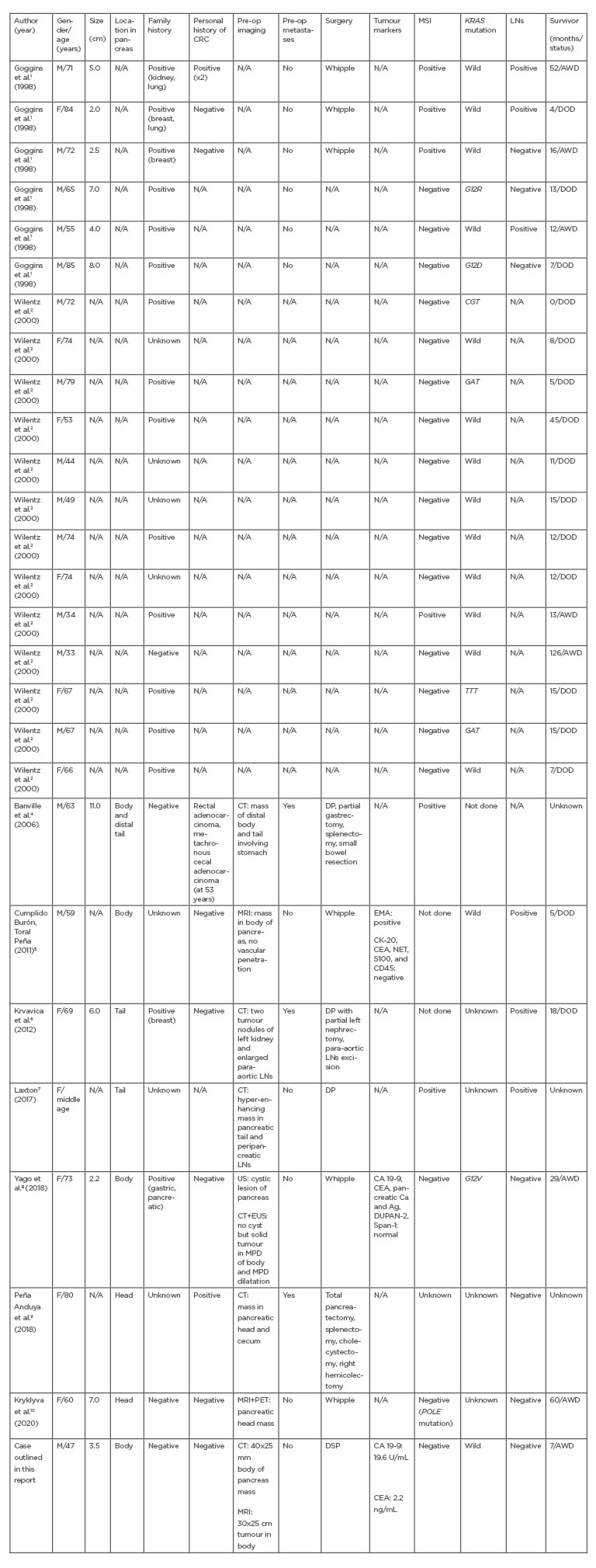
Table 1: Characteristics of previously reported medullary carcinoma of the pancreas.
Ag: silver; AWD: alive without disease; Ca: calcium; CA 19-9: carbohydrate antigen 19-9; CD45: leukocyte common antigen; CEA: carcinoembryonic antigen; CK-20: cytokeratin-20; CRC: colorectal cancer; DOD: died of disease; DP: distal pancreatectomy; DSP: distal spleen pancreatectomy; DUPAN-2: duke pancreatic monoclonal antigen Type 2; EMA: epithelial membrane antigen; EUS: endoscopic ultrasound; F: female; LN: lymph node; M: male; MPD: main pancreatic duct; MSI: microsatellite instability; N/A: not available; NET: norepinephrine transporter; Pre-op: pre-operation; ref: reference; S100: S100 protein; Span-1: S-pancreas-1 antigen; US: ultrasound; Whipple: Whipple procedure.
Medullary carcinoma of the pancreas is an exceptionally rare special subtype of pancreatic ductal carcinoma. The currently reported cases make it extremely difficult to elucidate the prognosis and optimal treatment for patients with medullary carcinoma of the pancreas. Finally, due to the rarity of this disease, combining the efforts of clinicians treating patients with medullary carcinoma of the pancreas through an international database would be of great benefit to patients.






