Abstract
von Willebrand disease (VWD) is considered the most common inherited bleeding disorder, even surpassing haemophilia A. Nevertheless, VWD may be underdiagnosed, overdiagnosed, or misdiagnosed, depending on the expertise of the managing clinician and the testing laboratory. This is due to the heterogeneity of VWD and the complexities of laboratory assessment. For example, in haemophilia, there is a deficiency or defect in a single clotting factor, either factor VIII or factor IX, for haemophilia A and B, respectively. In contrast, at least six types can be identified in VWD, classified according to the defect and/or deficiency in the plasma protein von Willebrand factor. von Willebrand factor has numerous functions, most of which can be assessed by laboratory testing. However, unlike haemophilia, a battery of laboratory tests is required to enable a diagnosis or exclusion of VWD, as well as its type classification. This complexity is not well understood by many clinicians or scientists. VWD was first described by Erik von Willebrand in 1926, and so 2025 represents 100 years of VWD. The authors review some of the history of VWD, as well as outlining the current state of play for diagnosis and treatment.
Key points
1. von Willebrand disease (VWD) is the most common inherited bleeding disorder, and is caused by a deficiency of, or defect in, the adhesive plasma protein von Willebrand factor.2. The first case of VWD was published in 1926, making 2025 its 100-year anniversary.
3. This review briefly describes the history of and summarises current strategies for VWD diagnosis and management.
INTRODUCTION
von Willebrand disease (VWD) is considered to be the most common inherited bleeding disorder, even surpassing haemophilia A. The estimated prevalence of haemophilia A, a sex-linked condition, is around one in 5,000 males, or around one in 10,000 people.1 The estimated prevalence of VWD is less clear. Using epidemiological data, or estimates based on patients providing test results potentially consistent with VWD, the estimated prevalence of VWD would be around one in 100 people (or up to 1% of the population).2,3 However, based on data reported to the World Federation for Hemophilia (WFH), the prevalence of VWD would also be around one in 10,000 people.1 Nevertheless, VWD may be underdiagnosed, overdiagnosed, or misdiagnosed, depending on the expertise of the managing clinician and testing laboratory.4 This is due to the heterogeneity of VWD, and to the complexities of laboratory assessment.
In haemophilia, there is a deficiency or defect in a single clotting factor, either factor VIII (FVIII) or factor IX (FIX), for haemophilia A and B, respectively. Accordingly, laboratories are in general able to assess for these clotting factors, or their deficiency, using standard haemostasis assays (typically one-stage assays, and occasionally chromogenic assays) on automated haemostasis analysers.5 Therefore, at least in developed countries, the reported number of haemophilia cases is close to 100% of the expected prevalence (Figure 1A). Nevertheless, in less developed countries, the reported number of haemophilia cases may be well below the expected prevalence.
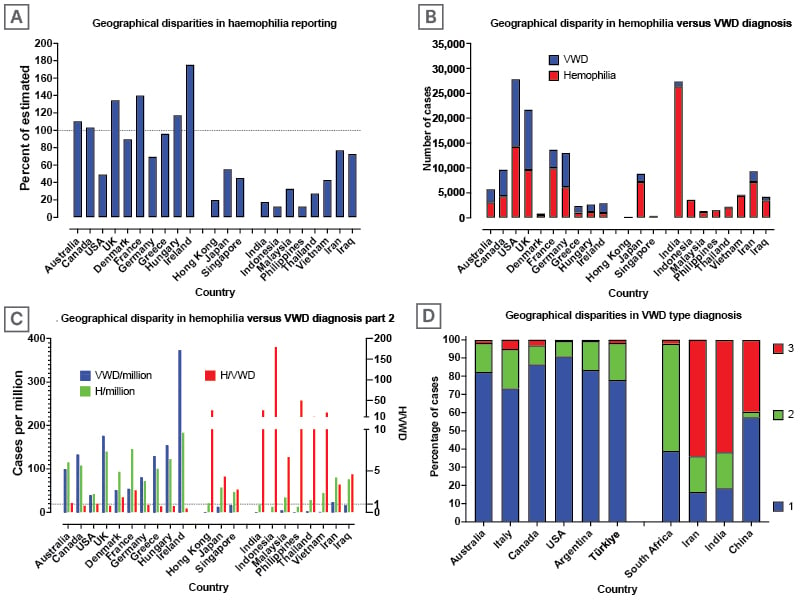
Figure 1: Geographical disparities in reporting cases of haemophilia and von Willebrand disease.
Data from WFH1 (Figures A, B, C) and Favaloro 20112 (Figure D)
(A) Geographical disparities in reporting cases of haemophilia. Close to 100% of estimated haemophilia cases are reported to the WFH from developed countries. Haemophilia cases seem to be underreported in some (e.g., developing) countries.
(B) Geographical disparities in reporting cases of haemophilia and VWD. Similar numbers of haemophilia and VWD are reported in most developed countries. Much fewer VWD cases than haemophilia cases appear to be reported in some (e.g., developing) countries.
(C) Data as per figure B, reported as cases per million population. Considering similar prevalence, the ratio of H/VWD should be close to one, which is the case for most developed countries. However, in some (e.g., developing) countries, the ratio exceeds 100, indicating a considerable underdiagnosis of VWD.
(D) Geographical disparity in VWD type diagnosis. Type 1 VWD is the most prevalent or reported VWD type in developed countries, and Type 3 VWD the least. In many developing countries, the situation is reversed, with Type 3 VWD the most prevalent or reported VWD type, and Type 1 VWD the least.
H/VWD: haemophilia/von Willebrand disease; WFH: World Federation of Haemophilia; VWD: von Willebrand disease.
In contrast to haemophilia, the divide in reported prevalence of VWD according to geographic jurisdiction is even more disparate.6 In most developed countries, the number of reported cases of haemophilia and VWD is similar (Figure 1B), resulting in an expected ratio of haemophilia/VWD of close to one (Figure 1C). However, this ratio exceeds 100 in some developing countries (Figure 1C), meaning that VWD is predominantly undiagnosed or potentially misdiagnosed in those countries. On the other hand, in some countries, the reported prevalence of VWD is higher than expected (Figure 1C), raising the possibility of overdiagnosis. Alternatively, this may reflect more accurate data collection, thereby again raising the possibility of underdiagnosis in other countries.
At least six types of VWD can be identified, with these classified according to the defect and/or deficiency in the plasma protein von Willebrand factor (VWF).7 However, VWD is conventionally divided into three major types: 1, 2, and 3. Type 1 VWD represents a deficiency of VWF, and thus is a quantitative disorder. The VWF that is present in plasma, albeit in reduced quantity, has similar function to that in individuals without the disease (i.e., Type 1 VWD does not generally represent a qualitative defect; a potential exception is a subtype subject to higher plasma clearance). Type 2 VWD is represented by qualitative defects in VWF, and thus represents qualitative disorders. VWF has numerous functions, most of which can be assessed by laboratory testing. Type 2 VWD is subclassified into four (sub)types: loss of high molecular weight (HMW) VWF (Type 2A VWD); heightened VWF activity leading to the clearance of HMW VWF and platelets from circulation (Type 2B VWD); loss of VWF ability to bind to FVIII, leading to clearance of FVIII from circulation (Type 2N VWD); and finally reduced VWF activity not associated to any of the above (Type 2M VWD). Type 3 VWD is the most severe, but globally the rarest form of VWD. It is characterised by a total deficiency of VWF production. Not only is there a geographic disparity in VWD versus haemophilia diagnosis, but there is also a geographic disparity in VWD subtype diagnosis (Figure 1D), with Type 1 and Type 3 VWD representing more straightforward to identify compared to Type 2 VWD. Type 1 VWD is the most commonly identified type, especially in developed countries,2 although it is also subject to overdiagnosis. In developed countries, the reported prevalence of Type 3 VWD is around one in one million people. In contrast, Type 3 VWD is often the predominant VWD type diagnosed in developing countries.2 This disparity may be due to an increased prevalence of Type 3 VWD in these countries, perhaps resulting from more frequent consanguineous relationships. Furthermore, since this type is the most severe, patients with Type 3 VWD are most likely to seek medical attention. Type 2 VWD is variously reported, as these diagnoses require multiple laboratory investigations, which are not always available in some countries.6
In summary, unlike haemophilia, a battery of laboratory tests is required to enable a correct diagnosis or exclusion of VWD, as well as to enable effective type classification.8,9 This complexity is not well understood by many clinicians or scientists. VWD was first described by Erik von Willebrand in 1926,10 and so 2025 represents 100 years of VWD. The authors review some of the history of VWD, and outline the current state of play for diagnosis and treatment.
A SHORT HISTORY OF VON WILLEBRAND DISEASE DIAGNOSIS
As noted, VWD was first described by Erik von Willebrand in 1926,10 although he termed it ‘hereditary pseudo-haemophilia’. He described an index case of a 5-year-old female he first assessed in April 1924, as well as an investigation of her extended family. The patient was admitted to hospital for investigation of severe bleeding from her nose and gums. Her parents were cousins, and there was a positive bleeding history within the family.11 Her siblings had a bleeding history, and three had died from gastrointestinal bleeds. In contrast to haemophilia, both sexes were affected, and mucosal bleeding represented the predominant symptom. A prolonged bleeding time with a normal platelet count was the most important laboratory abnormality identified, suggesting a functional disorder of the platelets associated with a systemic lesion of the vessel wall. Naturally, at that time, no tests for VWF or its many activities were available.
In the 1950s, after preliminary methods were established to measure FVIII (then called anti-haemophilic factor), several clinicians described patients of both sexes who had prolonged bleeding times associated with reduced FVIII.11 These investigators proposed that these patients were deficient in a factor other than that responsible for classic haemophilia A. It was not until 1971 that it was understood that the deficiency of a new factor, later recognised to be VWF, and different from FVIII, was actually responsible for the disease.11
An antiserum against a highly purified preparation of FVIII was developed by Zimmerman’s group in the early 1970s, and then used to identify this new plasma protein using an immunochemical technique.12 This protein, initially named FVIII-related antigen, and later understood to be VWF antigen (VWF:Ag), was present in individuals without the disease and in those with haemophilia A, but was lacking in patients with VWD. This immunologic technique became the cornerstone of laboratory testing that was able to distinguish patients and carriers of haemophilia from those with VWD.12 Around the same time, Howard and Firkin observed that an antibiotic called ‘ristocetin’ caused platelet agglutination in platelet-rich plasma from individuals without disease, but not in the platelet-rich plasma of patients with VWD.13 This ristocetin effect could be linked to the presence of the FVIII-related antigen (i.e., VWF:Ag). This development led to the subsequent use of the ristocetin-induced platelet agglutination/aggregation assay, as well as a quantitative assay for VWF activity called ristocetin cofactor (VWF:RCo).13,14 The dual application of these VWF:Ag and VWF:RCo assays using the plasma of patients with VWD permitted the identification of different VWD types during the late 1970s, in various countries, and by various workers in the emerging field of VWD diagnosis.11
In the early 1980s, Ruggeri and Zimmerman reported a pivotal observation of abnormal multimeric structure of VWF in different VWD variants.15 Many laboratories have since used this technique, as well as developing various improvements, to identify additional VWD variants characterised by the loss of HMW VWF multimers and abnormal VWF structure. The VWF gene was cloned in 1985, which led to increasing understanding of the molecular basis of VWD.11,16,17 In 1994, Sadler published the initial VWD classification, where Type 2 was divided into the four categories that are still used today: 2A, 2B, 2M, and 2N.18
Also in the 1980s came the development of additional VWF activity assays to supplement the original VWF:RCo assay. In 1986, Brown and Bosak first described the original VWF collagen binding assay (VWF:CB),19,20 and in 1989, an assay to identify Type 2N VWD was first described; this being the VWF FVIII binding (VWF:FVIIIB) assay.21
In the 1990s, the discovery of the VWF cleaving protease, ADAMTS13, was made.22,23 The authors can probably fast-track here to the new millennium, the 2000’s, where further refinements were made to various VWF activity assays, as well as to the standard VWF:Ag and VWF:RCo assays.24 First, VWF:Ag assays moved from immunological gel based procedures (time-consuming, complex, and unsuited to high throughput) to ELISA, permitting large-scale testing or patient screening. They then advanced to latex immunoassays (LIA) on automated haemostasis analysers, which now represents the predominant VWF:Ag methodology. Similarly, VWF:RCo moved from initial visual platelet agglutination to semi-automated methods using platelet aggregometers, and then to modern methods using automated haemostasis analysers. In addition, the standard VWF:RCo assay, using platelets, usually fixed or lyophilised, has morphed into more modern alternatives using inert particles (latex or magnetic beads) and recombinant glycoprotein Ib (GPIb), this being the platelet VWF receptor responsible for platelet agglutination/aggregation in the presence of ristocetin. These newer assays are called ‘VWF:GPIbR’ (R for recombinant), and can be run like LIA on automated haemostasis analysers, as well as by chemiluminescence immunoassay on an instrument called the AcuStar® (Werfen, Barcelona, Spain). Finally, a newer VWF activity assay uses gain-of-function GPIb mutations to enable agglutination of latex particles without the need for ristocetin. This assay, called ‘VWF:GPIbM’ (M for mutant recombinant), can also be run like LIA on automated haemostasis analysers.
The 2000s and 2010s also saw the development and refinement of standardised bleeding assessment tools to enable qualification and quantification of bleeding symptoms, which provide a clinical aid in assessing bleeding severity or risk.25-27
A SHORT HISTORY OF VON WILLEBRAND DISEASE MANAGEMENT
Needless to say, the management of VWD has similarly seen quite an evolution over the past 100 years. There was really no suitable treatment available for VWD until 1956, when a plasma fraction called ‘I-O’ was prepared by Blombäck and Blombäck.28 This fraction was able to correct both FVIII and bleeding time defects. A second advance was made when Pool and Robinson were able to demonstrate that a cold fraction of plasma cryoprecipitate contained anti-haemophilic factor, and could correct the bleeding defects in both haemophilia A and VWD.29 Cryoprecipitate was easily prepared by blood banks, and was thence extensively used for management of VWD, because it provided large amounts of VWF without causing volume overload. Cryoprecipitate may still be used in some countries worldwide, where specific VWF concentrates are not yet available, and remains on the WHO listing of essential medicines.30 However, a major disadvantage of cryoprecipitate is that virucidal methods cannot be applied to it, so it carries a small but real risk of transmitting blood-borne infections.31,32
In 1977, Mannucci et al.33 pioneered the use of desmopressin (DDAVP) into clinical practice, following the earlier work of Cash et al.34 DDAVP was effective in achieving haemostasis in patients with mild haemophilia and VWD who were undergoing surgery, without major side effects. The use of DDAVP can, in some circumstances, avoid the use of VWF/FVIII concentrates, and probably prevented many cases of viral infection in the early 1980s.11
The development of clotting factor concentrates became increasingly successful, providing increasing purity and safety, with various virucidal methods applied. Therefore, virus-inactivated FVIII/VWF concentrates, originally developed for the treatment of haemophilia A, played increasingly important roles in the management of patients with VWD who were unresponsive to DDAVP. The first pasteurised FVIII/VWF concentrate, Haemate-P/Humate-P, became available in Germany in 1981.11 Many additional FVIII/VWF concentrates have since been developed. Using various purification and viral inactivation processes, all current FVIII/VWF concentrates are effective for VWD management. However, individual concentrates may differ in terms of relative levels of FVIII and VWF, as well as in their retention of HMW VWF.35 Indeed, some VWF concentrates are virtually devoid of FVIII, whereas other concentrates have similar proportions of FVIII and VWF. In 2025, these can be variously applied to different types of VWD, or at different periods of their management, according to the concept of personalised management.35,36
Another major advance in VWD management was the development and availability of a recombinant VWF concentrate.37 This concentrate does not contain FVIII, but expresses high levels of HMW VWF, since the material has never been exposed to ADAMTS13. Its use can be applied with or without additional (recombinant) FVIII, depending on VWD type and the clinical situation, again according to the concept of personalised management.35,36
There are additional (adjunct) therapies that may also be applied to individuals with VWD, as detailed in the section ‘MANAGEMENT OF VON WILLEBRAND DISEASE IN 2025’, as well as elsewhere.35
DIAGNOSIS OF VON WILLEBRAND DISEASE IN 2025
The diagnosis of VWD requires both clinical assessment and laboratory testing.8,9 For clinical evaluation, a physical evaluation is required, especially to assess for bruising, and a clinical history for the patient and extended family taken. The clinical history can be obtained using structured tools such as a bleeding assessment tool, or via standard consultation.
In 2025, well-equipped laboratories have an armamentarium of tools to assist the diagnosis or exclusion of VWD, as well as its correct classification.38,39 The main assays in use for this purpose are summarised in Table 1. Not all tests need to be performed on all patients under investigation for VWD. It is usual to start with a set of VWD ‘screening’ assays, namely FVIII:C, VWF:Ag, and one of the VWF GPIb binding (VWF:GPIbB) activity assays (i.e., VWF:RCo, VWF:GPIbR, or VWF:GPIbM).8,9 If all three tests are normal, and if the ratio of VWF:GPIbB/Ag is >0.7, then VWD can be excluded with a high degree of confidence. If all three tests are normal, but the ratio of VWF:GPIbB/Ag is <0.7, then Type 2 VWD cannot be excluded, and the patient should be further investigated.
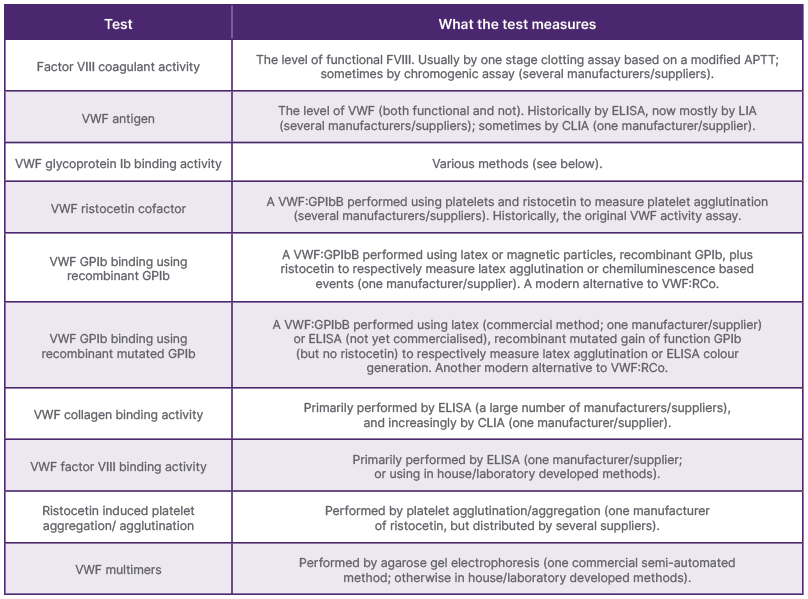
Table 1: Summary of the main tests used to diagnose/exclude von Willebrand disease.
APTT: activated partial thromboplastin time; CLIA: chemiluminescence immunoassay; FVIII:C: factor VIII coagulant activity; GPIb: glycoprotein Ib; LIA: latex immunoassay; RIPA: ristocetin induced platelet aggregation/agglutination; VWF: von Willebrand factor; VWF:Ag: von Willebrand factor antigen; VWF:CB: von Willebrand factor collagen binding activity; VWF:FVIIIB: von Willebrand factor VIII binding activity; VWF:GPIbB: von Willebrand factor glycoprotein Ib binding activity; VWF:GPIbM: von Willebrand factor glycoprotein Ib binding using recombinant mutated glycoprotein Ib; VWF:GPIbR: von Willebrand factor glycoprotein Ib binding using recombinant glycoprotein Ib; VWF:mult: von Willebrand factor multimers; VWF:RCo: von Willebrand factor ristocetin cofactor.
If one or more of the three tests are abnormal, but the ratio of VWF:GPIbB/Ag is still >0.7, then Type 1 VWD is possible. If one or more of the three tests are abnormal, but the ratio of VWF:GPIbB/Ag is <0.7, then Type 2 VWD is possible. If one or more of the three tests are abnormal, but the ratio of FVIII:C/VWF:Ag is <0.7, then Type 2N VWD is possible, although haemophilia A or a preanalytical issue is perhaps more likely. In any of these cases, additional investigation is required. In the case of possible Type 1 VWD, clinical history is key, since the new international guidelines suggest a diagnosis of Type 1 VWD in the case of VWF test results <30U/dL (or %), or where VWF test results are 30–50U/dL (or %) with a positive clinical history of bleeding. In these patients, it is also usual to repeat the basic test panel for confirmation, as VWF levels may fluctuate for a variety of reasons, for example, increasing during times of stress or after exercise. In the case of possible Type 2N VWD, this can be confirmed using the VWF:FVIIIB assay or genetic analysis of the VWF gene.8,9 Alternatively, repeat testing can be performed to exclude a preanalytical issue, given that FVIII is a labile coagulation factor and degrades quickly in plasma post-collection, or after plasma freeze-thaw events. Haemophilia A and Type 2N VWD should be distinguished, as different therapies are applied (FVIII concentrate for haemophilia A; VWF concentrate in Type 2N VWD). Clinical history, including family history and sex-linkage, will be useful to help distinguish them.
In cases where VWF:GPIB/Ag are <0.7, irrespective of whether the VWF tests themselves are normal or low, then Type 2A, 2B, or 2M is possible, as is the possibility of platelet type (PT) VWD, albeit rare.8,9 In these cases, clinical history is again important, as the presence of a family history suggests a congenital disorder, whereas a short personal history of bleeding/bruising, without a family history, may suggest an acquired condition. If VWF test results are normal, but VWF:GPIB/Ag is <0.7, then acquired VWD, Type 2B, or PT VWD are more likely than Type 2A or 2M VWD. If one or more VWF test results are abnormal, with VWF:GPIB/Ag <0.7, then Type 2A and 2M VWD are likely. In any of these situations, additional VWF tests are required for proper classification. In the case of possible Type 2B or PT VWD, the authors would reflex first to ristocetin-induced platelet aggregation (RIPA) assessment (with RIPA mixing if indicated), perform VWF:CB testing, and, only if required, conduct VWF multimer assessment.6,38,39 In the case of possible Type 2A or 2M VWD, the authors would still reflex first to RIPA assessment to exclude Type 2B or PT VWD, perform VWF:CB testing, and perform VWF multimer assessment to distinguish Type 2A from 2M VWD.
A flowchart of the authors’ diagnostic algorithm is provided in Figure 2. This algorithm differs from that recommended by the international guidelines,8,9 in that the authors employ the VWF:CB assay as a frontline assay, because they have previously identified several patients with VWD, especially those with Type 2M VWD,40 who would have been missed based on the standard three-test panel currently recommended by the international guidelines. This occurs due to assay variability and since VWF:GPIbB/Ag ratios are sometimes normal (i.e., >0.7) in Type 2M VWD.
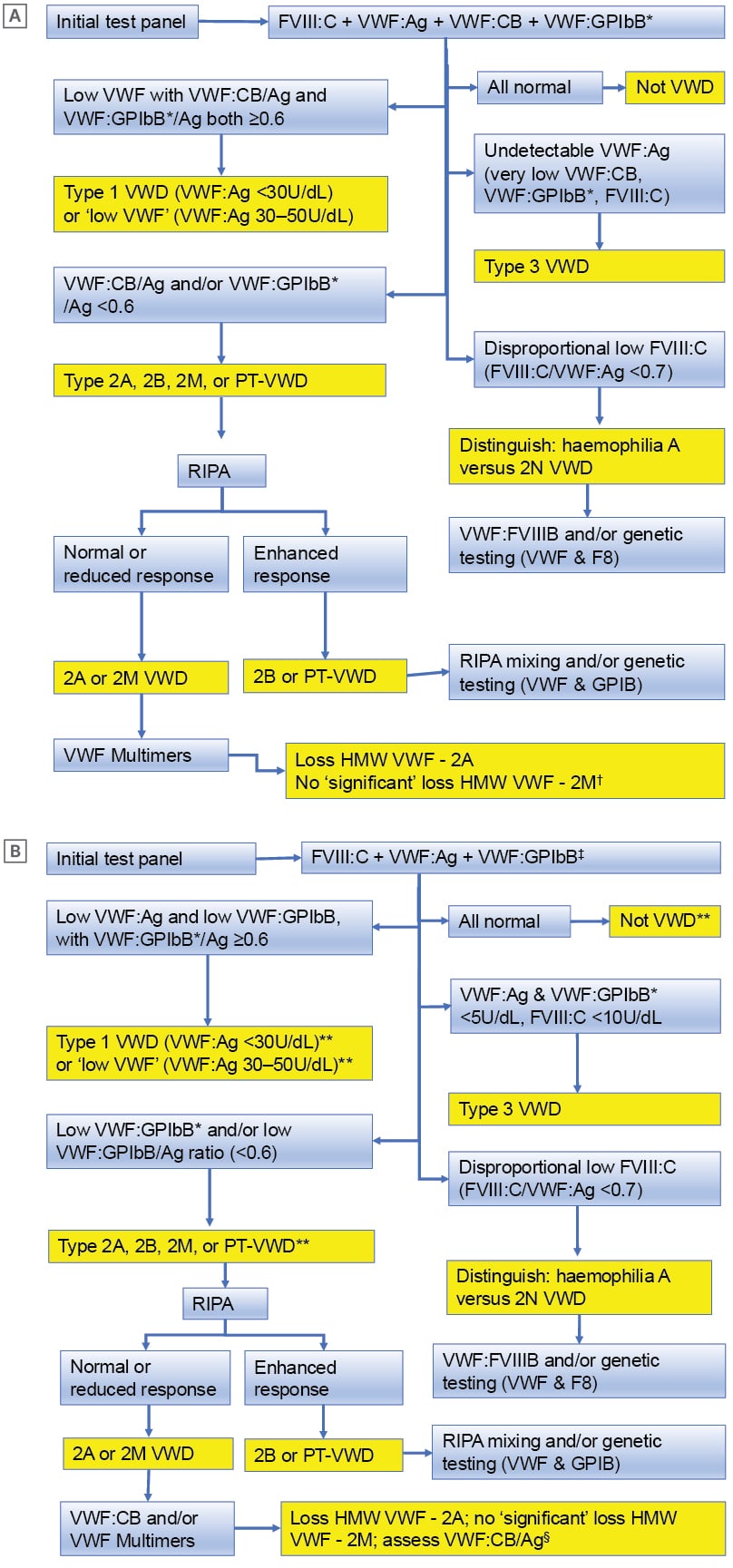
Figure 2: Algorithmic approaches to von Willebrand disease diagnosis or exclusion.
A) The approach taken in the authors’ laboratory using an initial 4-test panel. B) A potential alternative approach where laboratories are restricted to use of an initial 3-test panel.
*VWF:GPIbB = GPIb binding = VWF:RCo or VWF:GPIbR or VWF:GPIbM
?Usually VWF:GPIbB*/Ag <0.6 but VWF:CB/Ag ≥0.6 (2MGPIb); sometimes VWF:CB/Ag <0.6 and VWF:GPIbB*/Ag ≥0.6 (2MCB); sometimes VWF:CB/Ag and VWF:GPIbB*/Ag both <0.6 (2M)
‡VWF:GPIbB (GPIb binding; VWF:RCo or VWF:GPIbR or VWF:GPIbM)
§Repeat using fresh sample for confirmation (if relevant, exclude artifactual increase in VWF due to inflammation, infection, anxiety, stress, pregnancy)
**For 2M: usually VWF:GPIbB*/Ag <0.6 but VWF:CB/Ag ≥0.6 (2MGPIb); sometimes VWF:CB/Ag <0.6 and VWF:GPIbB*/Ag ≥0.6 (2MCB); sometimes VWF:CB/Ag and VWF:GPIbB*/Ag both <0.6 (2M)
FVIII:C: factor VIII coagulant activity; GPIb: glycoprotein Ib; HMW VWF: high molecular weight von Willebrand factor; PT-VWD: platelet type von Willebrand disease; RIPA: ristocetin induced platelet aggregation/agglutination; VWF: von Willebrand factor; VWF:Ag: von Willebrand factor antigen; VWF:CB: von Willebrand factor collagen binding activity; VWF:FVIIIB: von Willebrand factor VIII binding activity; VWF:GPIbB: von Willebrand factor glycoprotein Ib binding activity; VWF:GPIbM: von Willebrand factor glycoprotein Ib binding using recombinant mutated glycoprotein Ib; VWF:GPIbR: von Willebrand factor glycoprotein Ib binding using recombinant glycoprotein Ib; VWF:mult: von Willebrand factor multimers; VWF:RCo: von Willebrand factor ristocetin cofactor; VWD: von Willebrand disease.
Adapted from Favaloro and Pasalic6
MANAGEMENT OF VON WILLEBRAND DISEASE IN 2025
The authors have previously outlined their management approach for VWD.35,41 In brief, they would undertake a DDAVP trial for most patients with VWD to assess effectiveness, as this differs on a patient-by-patient basis. In the authors’ geographic locality, recombinant VWF is not available, and so they would apply their locally available VWF/FVIII concentrate, which is Biostate® (CSL Behring, Broadmeadows, Australia) as required based on the patient’s VWD type and situation. Additional therapies, including hormonal agents, and the anti-fibrinolytic agent tranexamic acid, may be used in select situations. The authors’ approach would be similar to that applied in other geographic locations, although Biostate will be substituted with recombinant VWF, or other locally available VWF/FVIII concentrates. In some locations, nasal DDAVP is available, and in others, aminocaproic acid may be used in place of tranexamic acid.
WHY DOES VON WILLEBRAND DISEASE REMAIN UNDERDIAGNOSED, OVERDIAGNOSED, AND MISDIAGNOSED IN 2025?
So, given the armamentarium of tests available to everyone in 2025, why is VWD still underdiagnosed, overdiagnosed, or misdiagnosed in 2025? Underdiagnosis is the most likely scenario.4 As indicated in the introduction, VWD is underdiagnosed in many countries (Figure 1), most likely because incomplete test panels are employed (e.g., only FVIII:C or VWF:Ag; or only FVIII:C and VWF:Ag). VWD, in particular Type 2 VWD or Type 2B or PT VWD, may be underdiagnosed because in many cases, VWF tests results are in the normal range. The clue for these may be the reduced VWF:GPIbB/Ag ratio, but its significance missed.
Misdiagnosis of VWD is the second most common scenario.4 Type 3 VWD is sometimes misdiagnosed as haemophilia A. This will occur when patients are only assessed for FVIII:C, and VWF testing is not performed. Type 2N VWD is also often misdiagnosed as haemophilia A. This is because haemophilia A is more common than Type 2N VWD, and the possibility of 2N VWD is not considered, especially in male patients. In particular, access to genetic testing for the VWF gene or to the VWF:FVIIIB assay is not readily available, especially in developing countries.6
Overdiagnosis of VWD is also possible, especially for Type 1 VWD. A reduced level of VWF does not, on its own, confirm a diagnosis of VWD; there also needs to be a positive clinical history.8 Incorrect diagnosis of VWD can also occur due to preanalytical issues, which may yield false low levels of VWF.42 Here, repeat testing for confirmation is critical. Individuals with blood group O have lower levels of VWF than those non-blood group O, and are therefore at greater risk of overdiagnosis.4
CONCLUSION
The authors provide an updated and concise review of the diagnosis and management of VWD, with reference also to its preceding history in this, the 100th anniversary of VWD. VWD remains underdiagnosed, overdiagnosed, or misdiagnosed in 2025, due to a variety of factors, including limitations in the test panels used, lack of recognition regarding test patterns, and the limitations of various VWF tests. VWD diagnosis can be improved by education and by increasing the number of tests used for any given patient, including the inclusion of a VWF:CB assay. VWF assays have increasingly improved over the years, including the development of more modern alternatives to VWF:RCo. These refinements will continue to develop over the next 100 years of VWD history. The management of VWD will also improve, as recombinant VWF becomes increasingly deployed worldwide and is used to tailor individual therapy according to the VWD type and clinical situation, under the concept of personalised therapy.






