
Abstract
Multiple myeloma is increasingly being recognised as more than one disease, characterised by marked cytogenetic, molecular, and proliferative heterogeneity. The prognosis is widely varied, ranging from low to very high-risk, based on cytogenetic and molecular studies. Although novel agents, such as proteasome inhibitors and immunomodulators, have been developed, which have improved treatment responses and disease prognosis, multiple myeloma remains an incurable disease. Based on highly sensitive detection tools, such as gene expression profiling and next generation sequence analysis, and the understanding of the pathogenesis of multiple myeloma, many potential agents, including monoclonal antibodies, drug-conjugated antibodies, drugs targeted to molecular abnormalities, microRNA inhibitors or mimics, and immune therapies, such as chimeric antigen receptors T cells and anti-PD1 agents, can be considered personalised therapies. In this paper, multiple myeloma pathogenesis and potential molecular and immunotherapies are reviewed.
INTRODUCTION
Multiple myeloma (MM), which accounts for approximately 10% of all haematological malignancies, is a clonal, late B cell malignancy in which plasma cells (PC) accumulate in the bone marrow (BM).1 The disease undergoes multistep transformations and its genetic landscape changes over time owing to additional events, such as somatic mutations and epigenetic and chromosomal copy-number changes; this drives the progression of MM from monoclonal gammopathy of unknown significance to symptomatic MM, often leading to an aggressive, extramedullary disease.2 Although many novel therapeutic agents have been developed, MM still remains an incurable disease, possibly owing to its marked genetic heterogeneity.
Recently, a range of sensitive detection tools have been developed, including analysing cytogenetic abnormalities in M-phase cells and interphase fluorescent in situ hybridisation; molecular analysis using a microarray, which has identified a 70-gene signature of an aggressive form of the disease (gene expression profile [GEP]70);3 and next generation sequence analysis. However, further investigation is required to understand the pathogenesis and target genes of MM to determine the best treatment strategies for each patient.
PATHOGENESIS OF MULTIPLE MYELOMA
There are two key aspects of MM pathogenesis: the genetic lesions intrinsic to the malignant clone, and the interaction between myeloma cells and the microenvironment of the BM.4
Genetics of the Myeloma Cell
MM is classified as either nonhyperdiploid or hyperdiploid based on karyotype analysis. The hyperdiploid subtype accounts for 50–60% of MM patients5 and is characterised by the presence of copy-number alterations such as trisomies of the odd chromosomes (3, 5, 7, 9, 11, 15, 19, and 21).4 Nonhyperdiploid subtypes harbour translocations between 14q32, immunoglobulin heavy chain locus, and one of several partner oncogenes, including MAF (16q23), MAFB (20q11), FGFR3/MMSET (4p16.3), Ccnd1 (11q13), and Ccnd3 (6p21).6
Hyperdiploid Molecular Pathway
There are approximately 35 nonsynonymous mutations per MM case.7 Interstitial copy-number gain associated with increased gene expression or with activating mutations in oncogenes represents another set of drivers for MM progression. For example, the amplification of 1q potentially involves more than one relevant oncogene, including CKS1B, ANP32E, BCL9, and PDZK1.8 Interstitial copy-number gains resulting in amplification of NIK (Map3k14), TAC1 (Tnfrsf13b), and LTBR proteins can also activate the nuclear factor (NF)-κB pathway.9
Nonhyperdiploid Molecular Pathways
Deregulation of the G1/S Phase transition is a key early molecular abnormality in MM. Consistent deregulation of a D-group cyclin was first noted whilst studying the t(11;14) and t(6;14) translocations, which deregulated CCND1 and CCND3, respectively.10 MAF (t[14;16]) and FGFR3/MMSET (t[4;14]) upregulate CCND2 and deregulate G1/S transition.10
Upregulation of NF-κB signalling is important in MM. FAM46C, DIS3, CYLD (16q), BIRC2 (also known as CLAP1), Birc3 (11q), and Traf3 (14q32) are all tumour suppressor genes involved in the NF-κB pathway,7-9,11 and are inactivated in MM. Furthermore, 58% of MM cases show loss of a negative cell cycle regulator, downregulation of Cdkn2c by loss of chromosome 1p32, silencing of Cdkn2a by methylation,8,12 and inactivation of Rb1 as a result of loss of chromosome 13. LTB, a Type II membrane protein of the tumour necrosis factor (TNF) family, heterodimerises with LTA to generate the ligand for LTBR, which is a positive regulator of the NF-κB pathway frequently amplified in MM.13
The MAPK/ERK pathway is frequently deregulated, induced by genes, including NRAS in 24%, KRAS in 27%, and BRAF in 4% of MM patients.14 Deregulation of the PI3K/AKT pathway, which was detected in 50% of cases, and the DEPTOR, a positive regulator, are also important.7,15 Loss of function of Tp53, the key gene at the 17p deletion site, is involved in the PI3K/AKT pathway and contributes to late oncogenesis of MM.16
The balance between apoptosis and antiapoptosis pathways contributes to cancer cell survival. t(14;18) involving the Bcl-2 locus was reported in 2–3% of MM patients, with more frequent protein level elevation.16 Bcl2l1 and Mcl-1 are similarly upregulated in MM via the interleukin (IL)-6/STAT3 axis.16 X-box binding protein 1 (XBP-1), a key molecule in guiding commitment and differentiation of PC, had dual roles in MM. Elevation of mRNA levels and spliced XBP-1 correlated with poor prognosis in MM.17 However, lack of spliced XBP-1 mRNA was shown to allow cells to escape from proteasome inhibitor-induced death and cause treatment failure and disease relapse.18 Sp140 showed truncating and missense mutations in MM.14 ROBO1, a transmembrane receptor implicated in β-catenin and MET signalling, harbours a truncating mutation, and Egr1, which encodes the early growth response one transcription factor, carries missense mutations in MM.
Epigenetics in Multiple Myeloma
Methylation of cytosines embedded in CpG islands in the promoter region of target genes is an epigenetic mechanism of transcriptional silencing. A number of tumour suppressor genes are hypermethylated early in MM, and increased gene methylation occurs during the process of MM progression.19 Histone methylation and acetylation are also altered in MM.20 The most pronounced DNA methylation change is seen in the 15% of patients with the t(4;14) translocation: these patients have increased gene-specific hypermethylation compared with other cytogenetic subgroups.20 MMSET, which is overexpressed in this t(4;14) subgroup, mediates histone 3 lysine 36 dimethylation. The dimethylation modification leads to deregulation and global changes in histone modifications that promote cell survival, cell cycle progression, and DNA repair.21 Bromodomains exert an epigenetic function via their direct interaction with acetylated lysine residues. The bromo and extraterminal family of bromodomains was shown to induce MYC expression in MM.22
microRNA
Dysregulated micro (mi)RNA expression in MM cells is associated with cytogenetic abnormalities and correlated with gene-expression changes characteristic of MM genetic subtypes and has provided the rationale for the design of new therapeutic strategies to treat MM.1 Of the 464 miRNA analysed, 95 were shown to be overexpressed in patients with MM (Table 1).23 These abnormally regulated miRNA target genes that regulate the cell cycle, apoptosis, survival, and cell growth; for example, the miR-17-92 miRNA cluster regulates Bcl-2;24 miR-19a and miR-19b, which form part of the miR17-92 cluster, downregulate SOCS-1;24 miR-29b regulates MCL-1;25 miR-21 regulates STAT3 in an IL-6-dependent manner; and miR-125b regulates BLIMP1 and IRF4.26 The miR106b-5 cluster, miR-181a and b, and miR-32 are also reported to target PCAF, a gene involved in p53 regulation and impacts tumour growth.27 Following analysis of 365 miRNA and gene expression profiling in 60 newly diagnosed MM patients, significant deregulation of miRNA was noted in different cytogenetic subtypes.28 For example, miR-203 and miR-342, located at 14q32, were downregulated in MM cells with t(4;14), and miR-1 and miR-113a were upregulated in MM cells with t(14;16).28
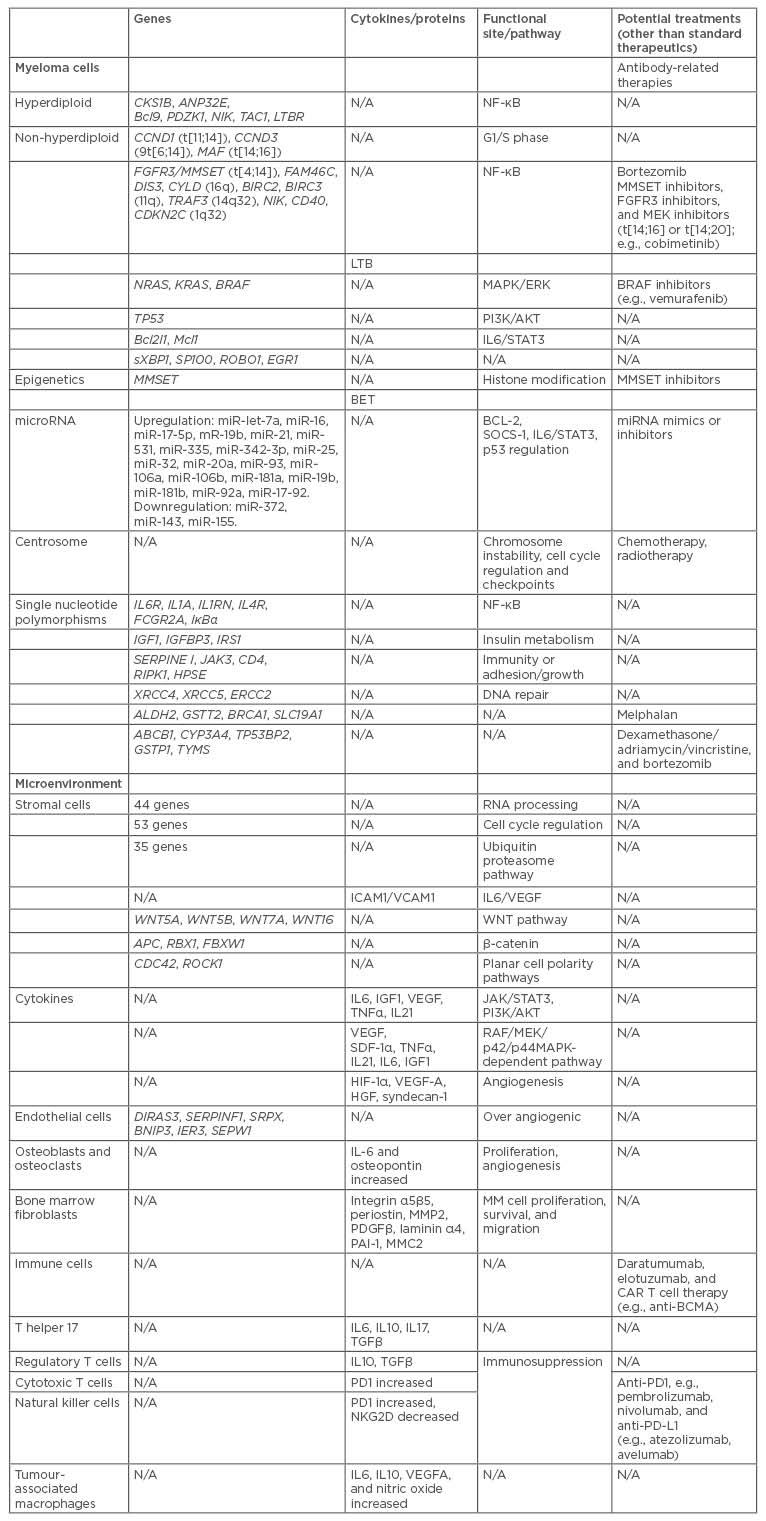
Table 1: Potential multiple myeloma treatments based on genetic mechanisms that promote disease progression.
BCMA: B cell maturation antigen; CAR: chimeric antigen receptors; CD: cluster of differentiation; IL: interleukin; miRNA: microRNA; MM: multiple myeloma; N/A: not applicable; NF-κB: nuclear factor κ B; t: translocation; TGFβ: transcription growth factor β; TNFα: tumour necrosis factor α; VEGF-A: vascular endothelial growth factor A.
Centrosomes
The function of the centrosome is to direct the mitotic bipolar spindles for accurate chromosome segregation during mitosis.29 Centrosome abnormalities in cancer correlate with chromosome instability and contribute to cell cycle regulation and checkpoints. Chromosomal instability and supernumerary centrosomes are typical in MM, representing early pathogenic events.
Single Nucleotide Polymorphisms
In MM and B cell development, highly polymorphic cytokine-encoding genes play an important role. When considering immune-related genes, an increased risk of MM was found to be associated with single polynucleotide polymorphisms (SNP) of IL6R, IL1A, IL1RN, IL4R, and FCGR2A,30 whereas SNP in IκBα, an inhibitor of the NF-κB pathway and the transcriptional activator TRAF3, showed protective roles.31 Other gene polymorphisms, including those in IGF-1, IGFBP3, and IRS1, related to insulin metabolism, also influence MM risk. Furthermore, polymorphisms in SERPINE I, JAK3, CD4, RIPK1, and HPSE immune-related and adhesion/growth genes have been associated with MM risk.32 SNP in genes related to DNA repair, including XRCC4, XRCC5, and ERCC2, showed controversial effects on MM risk.30
MICROENVIRONMENT OF MULTIPLE MYELOMA
Bone Marrow Stromal Cells, Cytokines, and Endothelial Cells
The BM microenvironment plays a critical role in the pathogenesis of MM. The close interplay between MM cells and the microenvironment, including BM stromal cells (BMSC), vascular endothelial cells, osteoclasts and osteoblasts, fibroblasts, and adipocytes, modulates and sustains growth, survival, and the development of drug resistance of MM cells. Several genes of BMSC, which have major functions in RNA-processing (44 genes), cell cycle regulation (53 genes), and ubiquitin proteasome pathway activation (35 genes), are dysregulated when cocultured with myeloma cell lines.33
The VLA-4 on MM cells binds to fibronectin in the serum, and the LFA-1 on MM cells binds to ICAM1 on BMSC,34 attracting MM cells to the BM. Other cytokines, such as TNF-α in the BM, can modulate the adhesion of MM cells by inducing NF-κB signalling. NF-κB-dependent upregulation of cell-surface adhesion molecules, such as ICAM1 and VCAM1, on both MM cells and BMSC, increases the binding capacity of tumour cells and BMSC, and induces the transcription and secretion of cytokines, such as IL-6 and vascular endothelial growth factor (VEGF) in BMSC.31 Cytokines in the BM microenvironment, such as IL-6, insulin-like growth factor 1 (IGF-1), VEGF, and TNF-α, mediate the growth of MM cells. IL-6, IGF-1, and IL-21 are associated with tumour-cell survival and resistance to apoptosis,35,36 which is mediated through the JAK/STAT3 and PI3K/AKT pathways. Other genes, including ANXA2P1 and ANXA2P2, are also related to myeloma cell adhesion and growth.37
The proliferation of MM cells is triggered by cytokines, such as IL-6, IGF-1, VEGF, TNF-α, stromal cell-derived factor-1α (SDF-1α), and IL-21, and is mediated through the RAF/MEK/p42/p44/MAPK signalling cascades.35,36 NRG3, expressed exclusively in myeloma cells and MM BMSC, is able to activate ErbB4 and promote myeloma proliferation.33 VEGF and SDF-1α, mediated through a protein kinase C and p42/p44/MAPK-dependent pathway, play important roles in cell migration. Wnt signalling, which is important in stemness, cell differentiation, and cell metabolism, is also deregulated in BMSC in MM patients. Upregulated expression of several non-canonical Wnt ligands (WNT5A, WNT5B, WNT7A, and WNT16), and upregulated expression of negative regulators of β-catenin (APC, RBX1 and FBXW1), would promote β-catenin ubiquitination for proteasome degradation.33 Conversely, the upregulated expression of some members of the non-canonical Wnt/Ca2+ and planar cell polarity pathways (e.g., CDC42 and ROCK1) may indicate a possible enhancement of the migration and invasiveness properties in MM patients.38
Constitutive activation of HIF-1α and aberrant expression of HIF-2 by MM cells elevated levels of VEGF-A, hepatocyte growth factor, and syndecan-1, which are seen in the BM microenvironment and contribute to angiogenesis.39 DIRAS3 (a GTP-binding RAS-like protein), SERPINF1, SRPX, BNIP3, IER3, and SEPW1 are correlated with the over-angiogenic phenotype of MM endothelial cells in active disease.40
Bone Marrow Niches
In addition to BMSC, osteoblastic and osteoclastic niches contribute to MM cell proliferation and angiogenesis through secretion of IL-6 and osteopontin.16
BM fibroblasts from MM produce ECM proteins, including integrin α5β5, periostin, MMP2, PDGF β, laminin α4, plasminogen activator inhibitor-1, lysyl-hydroxylase 2, prolyl 4-hydroxylase 1, nidogen-2, c-type mannose receptor-2, and basigin.41 Both BM fibroblasts and adipocytes are shown to support MM cell proliferation, survival, and migration.
Immune Cells
Immune cells show important supportive roles in MM. T helper 17 cells are abundant in the BM, under the priming of elevated concentrations of IL-6 and transcription growth factor-β, which suppress cancer immune surveillance by secreting IL-17 and IL-10. Increased proportion of functional regulatory T cells in the peripheral blood of MM patients directly correlates with worse prognosis.42 Cytotoxic T cells showed increasing expression of PD-1, a T cell receptor coreceptor with inhibitory function, in MM BM.43 Natural killer (NK) cells are also functionally impaired in MM patients with downregulation of the NK group 2D activating receptor and upregulation of inhibitory coreceptor PD-1. Type I NK T cells, expressing an invariant TCR, have been reported to play an important role in anticancer surveillance, and Type I NK-T deficiency was associated with MM progression and relapse.44 Tumour-associated macrophages contribute to MM pathogenesis in three ways: as a major source of IL-6; by producing IL-10, a major mediator of cancer immune tolerance by suppressing the function of T cells; and by releasing VEGF-A and nitric oxide.45
RISK STRATIFICATIONS
Risk category is based on the biological and molecular profile that predicts prognosis and treatment responses. Hyperdiploid myeloma and t(11;14) confer relatively favourable prognoses. MAF (t[14;16]), MAFB, or FGFR3/MMSET (t[4;14]) activation and deletion on chromosome 13 and/or 17 are associated with poor prognosis.6,46 GEP70 is used to identify high-risk disease and 10 subgroups are mentioned.3 Revised International Staging System (ISS) analysed 3,060 newly diagnosed MM patients.46 Based on ISS stages (β2-microglobulin and albumin), chromosome abnormalities (CA) and lactate dehydrogenase levels, the risk was categorised into three groups, from low-risk revised-ISS group I with ISS Stage I; no high-risk CA (del[17p] and/or t[4;14 and/or 14;16]) and normal LDH level; to high-risk R-ISS group III with ISS Stage III and high-risk CA or high LDH level.46
Mayo Stratification for Myeloma and Risk-Adapted Therapy divided active MM into three groups: high-risk, including del(17p), t(14;16), t(14;20), and GEP high-risk signature; intermediate risk, including t(4;14), del(13), hypodiploidy, and PC labelling index ≥3%; and standard risk, including t(11;14), and t(6;14). Patients with t(4;14) were shown to be at an advantage with bortezomib treatment.47
In cells with centrosome abnormalities, the threshold of apoptosis activation induced by drugs or radiation may be much lower than that in other cells.29 This may be because the functions of centrosomes are upstream of mitochondrial proteins (such as cytochrome c), Bcl-2 family proteins, and other apoptosis molecules such as Blc homology domain 3-only proteins and caspase 8, Noxa, TR3, BAK and BID, and TP53, which play an important role in cell cycle regulation and apoptosis.
TREATMENT
Current Treatment
Based on patients’ age, performance status, and other comorbidities, individuals with MM can be divided into autologous haematopoietic stem cell transplantation (ASCT)-eligible and ASCT-ineligible groups. Choosing the optimal initial therapy in MM remains a challenge. There are currently at least five classes of active agents available for the treatment of myeloma: alkylating agents (melphalan and cyclophosphamide), anthracyclines (adriamycin®, Pfizer, New York, New York, USA, and liposomal doxorubicin), corticosteroids (dexamethasone and prednisone), immunomodulatory drugs (thalidomide and lenalidomide), and proteasome inhibitors (bortezomib, carfilzomib, and ixazomib). Mayo Stratification for Myeloma and Risk-Adapted Therapy guidelines47 suggest four cycles of bortezomib, lenalidomide, and dexamethasone before ASCT, followed by ASCT and lenalidomide maintenance therapy, or, alternatively, tandem ASCT and bortezomib-based maintenance therapy, in standard and intermediate-risk patients, respectively. For high-risk patients, four cycles of carfilzomib, lenalidomide, and dexamethasone therapy, followed by ASCT or tandem ASCT and carfilzomib or bortezomib-based maintenance therapy for 2 years, was recommended. For ASCT-ineligible patients, bortezomib, lenalidomide, and dexamethasone therapy is suggested for all risk groups for 1 year and followed by lenalidomide and dexamethasone therapy in standard risk patients, and bortezomib-based maintenance therapy for a minimum of 1 year for intermediate or high-risk patients. In standard-risk patients >75 years old, lenalidomide and dexamethasone therapy was suggested as an initial treatment. However, this guideline has never been tested in prospective clinical studies to show superiority over other non-preferred regimens.
Molecular-Based Therapy
MM cells characterised by gene alteration involved in TNF/NF-kB signalling and antiapoptosis showed better responses and longer progression free survival (PFS) with bortezomib use.48 MM patients with t(4;14) (overexpression of MMSET and FGFR3) have better overall survival (OS) with bortezomib treatment.49 MMSET, FGFR3, and MEK inhibitors could be potential treatment choices in patients with t(14;16) or t(14;20). BRAF Val600Glu mutation, accounting for 4% of MM patients, is the target of BRAF inhibitors.49 To treat patients with unfavourable GEP, novel inhibitors (e.g., AURKA inhibitors) target overexpressed genes.49
SNP in MM cells also contribute to treatment responses. The TNFα-238A allele was correlated with prolonged PFS and OS in patients treated with thalidomide and dexamethasone.50 Treatment response of melphalan was associated with SNP in GSTP1, ALDH2, GSTT2, BRCA1, and folate transporter SLC19A1. Polymorphisms in ALDH2 and CYP1A1 were correlated with prolonged OS.51 Treatment response of dexamethasone/adriamycin/vincristine was correlated with SNP in ABCB1, CYP3A4, TP53BP2, GSTP1, and thymidylate synthase.52 T allele polymorphism in ABCB1 has been shown to have a better response to dexamethasone/adriamycin/vincristine and bortezomib treatment, as well as better PFS and OS.53 The poor metaboliser phenotype of CYP2C19 was associated with a poor response to thalidomide and increased risk of peripheral neuropathy.54
Other therapeutic approaches have been demonstrated in preclinical studies by investigating the activity of miRNA mimics or inhibitors on tumour cells. Certain miRNA showed increasing drug sensitivity, including LNA-i-miR-221 to melphalan, miR-2012 mimics to bortezomib, miR-150-5p mimics to glucocorticoids, and miR-29b to bortezomib, carfilzomib, and ixazomib.55
Antibody-Related Therapies
Several antigens that exhibit strong expression in MM cells, including cluster of differentiation (CD) 38, CD138, CD56, CD74, CD40, IGF-1R, SLAMF7, and immunoglobulin superfamily member FcRL5, may be candidates for antibody-related immunotherapy.56 Daratumumab and SAR650984 are anti-CD38 monoclonal antibodies that have shown satisfactory response rates in patients with relapsed/refractory MM and CD38+ haematological malignancies in separate Phase I clinical trials. A Phase III clinical trial of daratumumab, bortezomib, and dexamethasone, compared with bortezomib and dexamethasone, showed better overall response rate (ORR) (82.9% versus 63.2%) and better 1-year PFS (60.7% versus 26.9%) in 498 relapse/refractory MM cases.57 However, daratumumab monotherapy showed a reduced effect with an ORR of 31.1% in relapse/refractory MM.58 This reduced ORR may be a result of the enrolled patients having a higher refractory rate to last line of therapy compared to previous studies (62.2% versus 30.3%), which may reflect higher clonal heterogeneity, and missing stimulatory component of immunomodulatory drugs (IMiD) and bortezomib.
Elotuzumab is an antisignalling lymphocyte activating-molecule F7 monoclonal antibody. The mechanism of action of elotuzumab includes mediating antibody-dependent cell-mediated cytotoxicity, enhancing NK cell cytotoxicity, and disrupting MM cell adhesion to BMSC.59 The combination of elotuzumab, lenalidomide, and dexamethasone yielded an ORR of 79% in patients with refractory/relapsed MM in a Phase III ELOQUENT-2 clinical trial.60 There are two Phase III studies in newly diagnosed MM patients: ELOQUENT-161 and GMMG HD6.62 In ELOQUENT-1, 375 patients received elotuzumab plus lenalidomide and dexamethasone, and 375 patients received the control treatment of lenalidomide and dexamethasone; the results of this trial have not yet been published. In the GMMG HD6 Phase III clinical trial, 516 newly diagnosed MM patients are to be enrolled, and it aims to compare bortezomib, lenalidomide, and dexamethasone, with or without elotuzumab, in consolidation and maintenance therapies; this trial is also ongoing.
Other potential monoclonal antibodies include milatuzumab (an anti-CD74 monoclonal antibody),63 dacetuzumab,64 and lucatumumab.65 The anti-CD40 monoclonal antibodies, siltuximab, targeting IL-6,66 showed partial response (PR) in refractory/relapse MM. All of the aforementioned monoclonal antibodies should be considered for further personal treatment.
Another type of antibody-related therapy is antibody-drug-conjugated therapy: e.g., indatuximab ravtansine (BT062), an anti-CD138 antibody-drug conjugate (ADC);67 lorvotuzumab mertansine, an anti-CD56 ADC;68 and milatuzumab, an anti-CD74 ADC with doxorubicin,63 which induced a response in relapse/refractory MM as a monotherapy or combined with other drugs. The anti-FcRL5 maytansine analogue, DM4, and monomethyl auristatin E;69 the anti-B cell maturation antigen (BCMA), C269, antibody conjugated to monomethyl auristatin F;70 and the anti-BCMA ADC GSK285791671 showed good response in preclinical studies.
Immunotherapy
IMiD, including thalidomide, lenalidomide, and pomalidomide, induce an immune response. The drugs inhibit TNF-α production and angiogenesis by blocking the angiogenic growth factors, including basic fibroblast growth factor and VEGF. Specifically, these agents trigger and enhance caspase-8-mediated MM cell apoptosis and enhance both caspase-8-mediated MM cell apoptosis, triggered by FAS or TRAIL, and caspase-9-mediated MM cell killing, triggered by dexamethasone. IMiD also block the induction of cytokines, such as IGF-1, and inhibit IL-6 and VEGF secretion triggered by MM cell adherence to BMSC. In addition, IMiD inhibit angiogenesis and augment NK cell activity against autologous MM cells.72 Several clinical trials have demonstrated the benefits of using regimens involving thalidomide or IMiD, including lenalidomide and pomalidomide, for MM treatment, particularly in combination with proteasome inhibitors.73-76 This combined therapy has become the standard regimen for MM treatment.
T cells redirected to specific antigen targets with engineered chimeric antigen receptors (CAR) are emerging as powerful therapies in haematologic malignancies. Garfall et al.77 reported a refractory MM patient who achieved complete response (CR) while receiving autologous transplantation followed by treatment with CTL019 cells, which consists of autologous T cells expressing a CD3-ζ/CD137-based anti-CD19 CAR from a lentiviral vector. BCMA, a member of the TNF receptor superfamily TNFRSF17, is virtually absent on naïve and memory B cells, but is selectively induced during PC differentiation where it supports humoral immunity by promoting the survival of normal PC and plasmablasts. Ali et al.78 reported the first in-human clinical trial of T cells expressing anti-BCMA CAR in 12 advanced, heavily pretreated MM patients. Among these patients, one achieved stringent CR, one achieved very good partial response (VGPR), two achieved PR, and 8 patients had stable disease.78
The immune checkpoint inhibitor, PD-1, is upregulated on the surface of activated T cells, and its ligands (PD-L1 and PD-L2) are expressed on the surface of antigen-presenting cells and tumour cells. Pembrolizumab is a monoclonal antibody against PD-1 that helps to restore antitumour immune surveillance. KEYNOTE-023 is a Phase I dose-escalation study evaluating the safety and efficacy of pembrolizumab in combination with lenalidomide and low-dose dexamethasone in patients with relapse/refractory MM.79 With a median follow-up of 9.7 months (range: 4.3–18.4), 76% (13/17) of the patients evaluated for efficacy in dose determination/confirmation responded to treatment, including 4 VGPR and 9 PR, with a median duration of response 9.7 months (0.0–16.7). Three patients (18%) had stable disease. M protein or free light chains were reduced in 94% of patients.80 Badros et al.81 reported a Phase II study of 48 relapsed/refractory MM patients with at least two prior lines of therapy, treated with pembrolizumab, pomalidomide, and dexamethasone. ORR of PR or better was observed in of 27 of 48 patients (56%): stringent CR (8%), normal CR (6%), VGPR (13%), and PR (29%). Of the 18 high-risk patients, ORR was 33% including VGPR (11%) and PR (22%); however, another anti-PD1 antibody, nivolumab, did not show objective responses in MM.82 This may be attributed to the fact that the mechanism of action of T cell activity against MM cells does not involve PD-1 interaction with PD-L1.
Unlike ASCT, allogeneic stem cell transplant is a potentially curative option in MM, especially for the high-risk subgroup, which has several advantages, including a tumour-free graft and the potential for sustained immune-mediated disease control. However, the role of allogeneic stem cell transplant is limited due to high treatment-related mortality with conventional myeloablative conditioning regimens and controversial benefit in reduced-intensity/non-myeloablative conditioning regimens.83 Vaccination with dendritic cell and tumour fusions following ASCT reported marked expansion of myeloma-specific T cells and cytoreduction of minimal residual disease.83 In a Phase II clinical trial, Rosenblatt et al.84 demonstrate that repeated immunisation with a dendritic cell and tumour fusion vaccine after ASCT improved clinical response and late response rate after ASCT. Lenalidomide was reported to promote T cell proliferation and augment response to myeloma-specific tumour vaccines.85
CONCLUSIONS
Because of heterogeneities and clonal evolution of MM, a full understanding of the genetics of myeloma and its integration with standard clinical prognostic information may help design specific trials and treatments, especially for high-risk patients. Molecular-based therapies targeting MM cells and microenvironment have been studied recently. Immunotherapies, including CAR-T and checkpoint inhibitors, have shown promising results in relapse and refractory MM, with some effects in high-risk patients. However, a real, personalised, approach is still far behind the medical community, as CAR-T approaches with current antigenic selections and the current constructs have not really produced reliable effects. In the future, drugs targeting molecular pathways and immune therapies will be important for personalised treatment.





