Abstract
Von Willebrand disease (VWD) is a bleeding disorder, resulting from a quantitative or qualitative defect in von Willebrand factor (VWF). A regulatory role for VWF in angiogenesis was postulated upon the clinical observation that qualitative or quantitative VWF defects are associated with the frequent occurrence of neoangiogenesis, particularly in the gastrointestinal (GI) tract. Vascular malformations of the GI tract are a cause of digestive bleeding in the form of either acute or chronic haemorrhage and represent a heterogeneous group of lesions, including angiodysplasias and telangiectasias. The management of these patients is challenging due to recurrent and severe episodes of GI bleeding. The mainstay of treatment of angiodysplasia is replacement therapy when this abnormality causes GI bleeding in patients with congenital VWD. When bleeding episodes recur frequently, regular prophylaxis should be implemented, leading to an acceptable degree of prevention of bleeding. The authors had a difficult experience in a 14-year-old adolescent with Type 3 VWD, who had presented with extremely serious recurrent bleeding secondary to duodenal angiodyplasia.
Key Points
1. Gastrointestinal (GI) bleeding from angiodysplasia is a well-known complication of Von Willebrand disease (VWD). While the aetiology of duodenal angiodysplasia is still not clear, it may be a congenital process with increasing incidence as age progresses due to VWF driving the formation of Weibel Palade bodies.2. The diagnosis and treatment of recurring gastrointestinal (GI) bleeding in Von Willebrand disease is challenging, and associated with significant morbidity, as demonstrated in this case of a 14-year-old with Type 3 VWD, who presented with recurrent bleeding secondary to duodenal angiodyplasia.
3. While acute management of GI bleeding has been successful with Von Willebrand factor replacement therapy, prophylaxis has been less effective for preventing recurrent GI bleeding. Surgical intervention may be necessary in extreme cases, with excision of the culprit lesion and anastomosis.
INTRODUCTION
Von Willebrand disease (VWD) was first described in 1926 by Erik von Willebrand when he cared for a family member with a severe mucocutaneous bleeding problem.1 Type 3 VWD is inherited in an autosomal recessive fashion, and is the least common (0.5–4 per million in developed countries) and most severe form of the disease.2,3 Vascular malformations have been documented in patients with VWD since the 1960s, when Armand Quick first reported the presence of dilated blood vessels or telangiectasias in the noses of several patients.4 This association between angiodysplasia and gastrointestinal (GI) bleeding in patients with VWD was first reported in 1976 by Ramsay et al.,5 when they described GI vascular dysplasia as a cause of persistently recurring melena in two patients with VWD.Angiodysplasia is most frequently located in the colon, but can also occur in the small bowel and upper GI tract.6,7 Aggravation to these vessels leads to severe, intractable bleeding that often results in anaemia, hospitalisation for transfusion of packed red blood cells, and a significant decrease in quality of life.8 However, most of these angiodysplastic lesions will never bleed.6
The authors report the case of a 14-year-old male with Type 3 VWD and previous hospital admissions for severe anaemia with no visible blood loss.
Material and Methods
A 14-year-old male, carrier of Type 3 VWD, with a history of surgery for Hirschprung’s disease, wearing an ileostomy, was admitted for treatment of several episodes of melena. On admission, the patient was found in average general condition with intense mucocutaneous pallor, with inspection of the ileostomy pouch showing red blood in the pouch approaching 200 cc, and tachycardia at 120 beats/min. The patient would have been transfused several times at the main place of residence.
Results
Several hospitalisations had occurred before at the paediatric hospital of his chief place of residence, where the teenager had benefited from the transfusion of several red blood cells (20 AU total) and substitution in Von Willebrand factor (VWF) without any origin of bleeding able to be identified.
First Hospitalisation in Paediatric Hospital
The patient was readmitted with complaints of fatigue and an haemoglobin level of 4 g/L. Examination of the ileostomy pocket visualised melena-type bleeding. During this admission, the patient was again treated with tranexamic acid, and with transfusion of red blood cells with replacement in VWF. A digestive endoscopy found a DIII seat of a flat relief lesion of 12 mm with sheet bleeding. An injection of 10 cc of serum with adrenaline, followed by the establishment of two clips, allowed the immediate stop of the bleeding.
Second Hospitalisation in Paediatric Hospital
The patient was readmitted with the same problem. Again, stabilisation occurred. The upper GI endoscopy had made it possible to find at the level of the fundus of the stomach a vascular ectasia with a detached adherent clot. After washing with argon and placement of a haemostatic clip, there was also at the level of the D3 a sessile formation of 15 mm in diameter, major axis seat of two clips in place with visualisation of sheet bleeding at the level of the base. An injection of adrenalised serum was performed with placement of three haemostatic clips, which made it possible to control the bleeding.
Third Hospitalisation in Belfort Hospital
Because of the persistent bleeding without an obvious focus, the patient was transferred to the authors’ hospital, at 4 g/dL. The authors proceeded to a transfusion packed with red blood cells, with injection of tranexamic acid and substitution by concentrated Willebrand factor (Wilfactin) at a dose of 40 IU/kg every 6 hours. Another endoscopy had been performed, finding at the level of the third part of duodenum D3 a flat relief lesion of 12 mm, with a surface layer with clips in place. An injection of 8 cc of serum with adrenaline followed by the release of two clips. Haemostasis was achieved. Faced with these failures in endoscopic treatment, and given the seriousness of the clinical symptoms, the authors decided, along with the paediatric surgery team, to operate on the patient. Chirurgical treatment was carried out with a digestive endoscopy in order to locate the bleeding. This was very laborious and lasted 10 hours, given the pathology itself and the current episode (Figures 1 and 2).
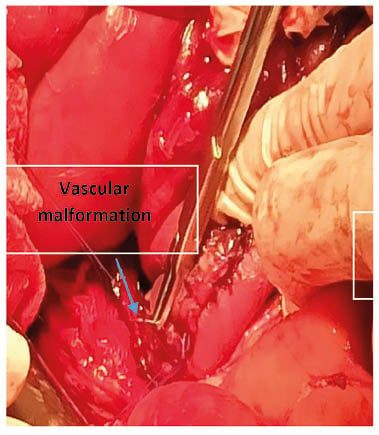
Figure 1: Location of the jejunal lesion.
The digestive endoscopy had made it possible to visualise a jejunal duodenum mucous seat due to vascular changes with diffuse inflammatory phenomena. A jejunal resection of 4 cm was carried out, and the part was sent for anatomopathological study. The microscopic study had shown an erosion of the mucous membrane by blood vessels; this mucosa and the submucosa are dilated, and sometimes dystrophic, with large haemorrhagic areas. This aspect is suggestive of vascular angiodysplasia. The postoperative follow-up was good, with a substitution in VWF in per postoperatively for 12 days, associated with tranxenamic acid.
The patient was discharged after 4 weeks of hospitalisation. They have not had any more bleeding after a follow-up of 10 months.
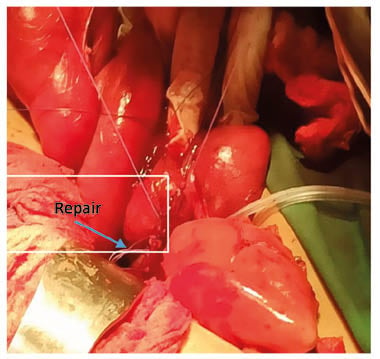
Figure 2: Resection anastomose.
DISCUSSION
The most common clinical manifestations of VWD include epistaxis, bruising, haematomas, and menorrhagia.9 GI bleeding from angiodysplasia is a well-known complication of VWD.9,10 In endothelial cells, VWF is constitutively synthesised and stored in Weibel Palade bodies 3.11,12 While the aetiology of duodenal angiodysplasia is still not clear, some theories were postulated to explain its pathogenesis in the view of its similarity with colonic angiodysplasia. Some considered angiodysplasia to be a congenital process in patients under 20 years old, with increased incidence as age advances, which may be attributed to VWF driving the formation of Weibel Palade bodies, the endothelial storage organelles that contain multiple proteins, including the angiogenesis regulator angiopoietin-2 (Ang-2).
Ang-2 is part of the Angiopoietins/Tie-2 pathway, a crucial system regulating vascular homeostasis and angiogenesis-89. Ang-2 has been shown to destabilise blood vessels and synergise with vascular endothelial growth factor to promote angiogenesis.14,15 VWF expression has been used extensively to quantify angiogenesis in a variety of tumours.16,17 However, VWF endothelial expression itself may be regulated in tumour endothelium. Authors reported up regulation of VWF expression by angiogenic factors: vascular endothelial growth factor and fibroblast growth factor 2,18 which are highly present in the tumour microenvironment. In its many interactions with an ever-growing list of molecules, VWF has come to be known as much more than a haemostatic protein.19 Angiodysplasia occurs most commonly in middle-age or elderly patients in the cecum and ascending colon, but also throughout the whole colon, small intestine, and stomach.10 The diagnosis and treatment of recurring GI bleeding in VWD is challenging, and associated with significant morbidity.20-22 Angiodysplasia is usually detected at endoscopy stage to evaluate GI bleeding but, in some cases, radiographic imaging or surgery may be required for detection.23,24
A number of alternative therapeutic measures have been attempted in the last few years for the management of recurrent GI bleeding in VWD.25 While acute management of GI bleeding has been successful with VWF replacement therapy, prophylaxis has been less effective for preventing recurrent GI bleeding.21,26
This patient had benefited from an endoscopic procedure with placement of clips with a replacement by Wilfactin and tranexamic, but after a short response time, this ended in failure. After several hospitalisations in a dramatic picture associating profuse bleeding and deglobulinisation, the authors opted for a surgical cure with excision of the lesion and anastomosis, which seems to have given in the authors’ patient with a follow-up of 10 months. Antifibrinolytic amino acids, such as tranexamic and epsilon aminocaproic acid, platelet concentrates, and combined oestrogen-progestogen drugs may be co-administered as adjuvants.27