Interview Summary
Lennox–Gastaut syndrome (LGS) is a severe childhood-onset epilepsy syndrome that can be challenging to identify due to its heterogeneous and evolving presentation. During interviews conducted by EMJ in July and August 2023, Karen Keough, a child neurologist specialising in paediatric refractory epilepsy at Texas A&M University School of Medicine, Round Rock, USA, and Rhys Thomas, an adult neurologist specialising in epilepsy (leading on learning disability and genetic testing) at the Royal Victoria Infirmary, Newcastle, UK, and Newcastle University, UK, gave their informed opinions on the challenges of diagnosing LGS. The experts spoke about the characteristic features of LGS, and how its gradual evolution from childhood onset can impede diagnosis. In particular, the issues around diagnosing LGS in adulthood, after considerable disease evolution and against a background of uncertain/incomplete medical history, were discussed. The experts also considered the 2022 International League Against Epilepsy (ILAE) position paper for the definition of epilepsy syndromes in childhood, and its potential impact for directing the diagnosis of LGS. Keough and Thomas concluded by describing how raised awareness, vigilance, and support in diagnosis could help to optimise treatment and readjust the goals for the management of adults and children with LGS.INTRODUCTION
LGS is a severe, refractory childhood-onset epilepsy syndrome. Clinical neurologists Keough and Thomas described how the variable, evolving presentation of LGS can create challenges in identification across the age spectrum from early childhood to adult years. As an overview, Keough explained: “LGS is a form of epilepsy that is very severe and refractory. It is heterogeneous, having a mixture of clinical presentation, and it is that variety that makes it more difficult to identify. It is clinically defined through a combination of clinical symptoms, medical history, underlying causes, progression and types of seizures, and electroencephalogram (EEG) findings.”1
Thomas added: “LGS is poorly understood by most people, and I think that is partly because there is no single cause, set of drug treatments, or outcome. We describe LGS as a network epilepsy, which means that something really potent happens during a critical time of development in a young child (it might be a genetic change, a tumour, something metabolic) that causes the brain to enter a pattern of seizures, and that pattern is recognisable no matter what the cause.”
Considering the array of symptoms in LGS, Thomas continued: “We see certain seizure types and patterns, and LGS almost always comes with an intellectual disability. In a normally developing infant or young person there may be regression (skill loss or arrestment) during times of high seizure frequency, so the seizures themselves contribute to the illness. People get tonic (stiffening) seizures, myoclonic (jumping) seizures, atonic (dropping) seizures.1 Sometimes you only get half the picture because the seizures are overnight, or there may be an obvious combination of seizures; for example, convulsive tonic–clonic [formerly ‘grand mal’] seizures, alongside atypical absences (blank spells). There is a real mix, which can also make it a challenge to treat.”
As the underlying pathology is linked to event(s) during early brain development, LGS has a characteristic childhood onset. There is no recognised adult onset, but Keough emphasised that the effects of LGS are not restricted to children: “This is a paediatric onset disease, but a lifelong condition. This is not something that remits, but it definitely evolves. From the time that the seizures begin, things are constantly changing but at a very slow pace, typically over years. In early childhood, seizures have a certain appearance with a mixture of different seizure types that become prominent; but then, as the brain changes, the types of seizures will wax and wane. So, LGS can look very different at one moment in time [compared with] a moment in time from years past. Indeed, the two presentations of epilepsy may look like they are not the same patient because there has been so much change, though it has been a gradual transition.”
Thus, for all the reasons outlined above, LGS is often unrecognised and unnamed, and Keough highlighted that someone affected by LGS may go through their whole life without a specific diagnosis beyond that of ‘epilepsy’.
THE IMPACT OF DIAGNOSIS
The experts were clear about the benefits of a prompt and specific diagnosis of LGS: for predicting disease evolution, handling expectations, and optimising treatment. Thomas observed: “If you are able to get an early ‘signature’ for likely LGS (for example, a characteristic EEG) as a paediatrician, you can then predict the limits; it might evolve, but in a predictable way. Whereas, if you are not proactive, it looks chaotic because every time there will be a new seizure type, a new reaction, a new problem with a drug.”
Being able to communicate this expected disease evolution to patients and their families is an important aspect of an early diagnosis. According to Keough: “It sets realistic expectations for caregivers and families, because many young children outgrow a tendency for seizures, but not if they have LGS. So, it is a different conversation, and it is a difficult conversation. They may have come with an expectation that the condition is going to get better, but this story is very different. Identifying LGS affects many things, but first and foremost it emphasises the need for a different philosophical approach to managing the patient. It generates a recognition that this is a refractory condition and so readjusts the goals. Day-to-day seizure activity is expected, so management is about trying to optimise care in order to minimise this activity, but it is also about avoiding over-treatment. Often in epilepsy, we are chasing the goal of complete seizure control, and can lose sight of the fact that treatments have consequences. Side effects may, for example, compound the patient’s cognitive impairment, and can really detract from quality of life.”
Thomas concurred: “If people with LGS have optimal seizure control, or an optimum combination of medicines, it is then about avoiding repeated trials of low-impact medicines that are never going to work, and which can cause patients and carers to lose faith in their doctor.”
However, reaching a diagnosis of LGS does more than point to treatment selection. As Keough explained: “A diagnosis is also important for getting access to medications. In the USA, if the patient does not have a diagnosis of LGS, insurance will not pay for the very expensive approved medicines.”
From a UK perspective, Thomas noted that although drugs licensed for LGS are centrally costed, it remains an intellectually rigorous process to secure a correct diagnosis, and select the appropriate licensed medication. Thomas concluded: “It is about prompt treatment with the right medicines, and the right strategy. Some of it is about daily medicine, some of it is about avoiding triggers, some of it is about emergency medicine benefits, but the quicker you know you are dealing with LGS, the quicker you can start to adequately treat the patient.”
INTERNATIONAL LEAGUE AGAINST EPILEPSY DIAGNOSTIC CRITERIA
The first ILAE position paper for the ‘classification and definition of epilepsy syndromes with onset in childhood’, including LGS, was published in 2022 (Table 1), and both experts heralded this as a significant advance for diagnosis.1 “For the very first time we are supported by guidance from the ILAE,” said Thomas. “It is an enormous piece of work, and I think it is practical, with a much broader voice than previous (well-respected) sources. They have generated guidance that makes sense in real life, giving you the features to look for, and also the features that are atypical. That is important, because not all tricky epilepsy is LGS,” Thomas said.
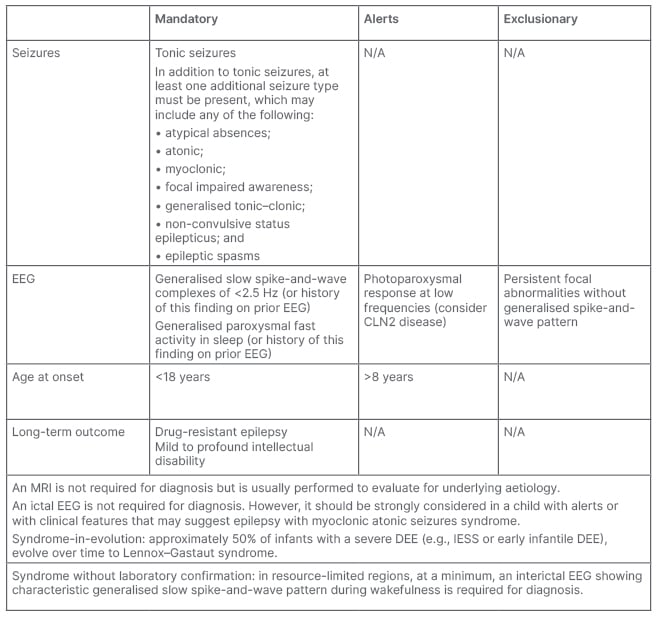
Table 1: International League Against Epilepsy (ILAE) diagnostic criteria for Lennox–Gastaut syndrome.
Alert criteria are absent in the vast majority of cases, but rarely can be seen. Their presence should result in caution in diagnosing the syndrome and consideration of other conditions.
This table was adapted from Specchio et al.1
CLN2: ceroid lipofuscinosis type 2; DEE: developmental and/or epileptic encephalopathy; EEG: electroencephalogram; IESS: infantile epileptic spasms syndrome; N/A: not applicable.
Keough agreed: “It is such a complicated syndrome to diagnose that people can get caught up in the details, and I think this particular layout of the diagnostic criteria is straightforward. It is also important to remember that not everything is going to be present in every patient. I like the way they have laid out the alerts [‘absent in the vast majority of cases but rarely could be seen’] and the exclusionary criteria.”
The experts then considered each aspect of the ILAE guidance for the diagnosis of LGS, centring on the triad of mandatory diagnostic features in turn: seizures, EEG, and age at onset (Table 1). Describing the seizure criteria, Keough stated: “’Tonic seizures’ are considered mandatory. However, the challenge is that sometimes you do not get a history of tonic seizures because they can be subtle and brief, and frequently occur only in sleep. So, video EEG monitoring is essential. Then, ‘at least one additional seizure type must be present’. I like that because it makes it easy to meet the bar of diagnosis, because LGS involves multiple seizure types. However, because this is a syndrome in evolution, what the [older] patient presents as their current seizure mixture probably is not what they have always had. You have to extract that from a history, which is going to have a variable degree of reliability.”
The EEG criteria (Table 1) were described as being well characterised and understood, but the experts also offered a caution regarding disease evolution, as well as the practicalities of resource use and availability. Thomas explained: “If you are a paediatrician, EEG gives you an early clue and you can pick out the characteristic features before you have had the full evolution of seizure types indicating LGS. This is great, but it makes more sense in a resource-rich situation and not in every healthcare setting. Also, young people with developmental issues may not like EEG on their head, even though it is an important part of the diagnostic process. In addition, some of the EEG features are not seen in adults, and if there has never been an EEG, or if notes are incomplete or lost, you do not have the evidence.”
Keough felt that a caveat in the guidance may be required to address this issue: “The EEG is helpful, but the criteria can be hard to elucidate because they are not always persistently present in older patients.2 That is the one issue that I would take with [the ILAE guidance]: the fact that these features might disappear is not addressed.”
Regarding the final element of the LGS triad, age of onset, the ILAE suggests a mandatory criterion for paediatric onset (<18 years), coupled with an alert for older cases >8 years, giving an element of flexibility. Keough felt this was fair for a condition that is so heavily early childhood onset.
Commenting on the practicalities of the ILAE guidance overall, Thomas remarked: “In terms of a consensus document, it is excellent. When I teach [the criteria], it is as traffic lights. A green is go [mandatory], red is a stop [exclusionary], and amber [alert] is just beware, and that is really helpful, because not every patient presents in the same way.”
DIFFERENTIAL DIAGNOSIS IN EARLY CHILDHOOD
The experts moved on to consider the specific challenges of diagnosing LGS in early childhood. According to Keough: “LGS is virtually never diagnosed at initial seizure onset. Even when acute brain injury, for example, is the cause, it takes time to evolve and meet the criteria that are required for diagnosis. Also, in the youngest patients the brain has not yet set up circuitry that is adequate to develop the electrographic features.” Thomas concurred, explaining that while LGS may be anticipated in a proportion of children, including those with a pre-disposing genetic syndrome (e.g., tuberous sclerosis), many patients lack a predictive aetiology, and it is most common to see someone slowly evolving to the full seizure repertoire. In this context, the experts emphasised that differential diagnosis of LGS from other severe infantile epilepsy syndromes can be an issue. Keough gave some examples: “A common scenario is a child who develops infantile spasms as their primary seizure type, but as their condition evolves, they develop other seizure types, and it becomes LGS. Then there are other very severe infantile epilepsies, such as severe myoclonic epilepsy in infancy or Ohtahara syndrome, where the infantile presentation does not initially fit the ILAE criteria for LGS, but in a few years they will get there. Also, many different kinds of genetic epilepsies could turn into LGS in some patients, though others will escape that path and avoid evolution to LGS.”
DIAGNOSIS IN ADULTS
The diagnostic difficulties outlined above can mean that LGS is not specifically identified until the adult years, which presents another distinct set of diagnostic challenges. Thomas explained: “As an adult neurologist, I see people with LGS who have had this syndrome for 20–30 years, by which time the disease evolution has probably plateaued. So, while paediatricians are trying to predict the future, we are trying to work out what happens at the beginning. It is two halves of the same journey, but with different challenges.”
Keough clarified: “It is not that the condition is new in adults (this is a developmental epilepsy that has been present from early childhood), it is exclusively because the diagnosis was missed when they were younger.”
According to Thomas: “There are lots of different ways to describe the same situation: some [neurologists] will be minimalistic and just say this is somebody with a learning problem and difficult-to-treat seizures; while others will describe the seizure types, the comorbidities, the aetiology; but then have not taken that big step back and added it all together. There might also be a piece of critical information missing. Certainly, before we had medicines licensed for LGS, there were lots of patients seen by paediatricians who were very comfortable diagnosing LGS, but had not put the name to it because it did not necessarily unlock or change anything. Whereas now, if you were to audit clinical records, I think you would see the phrase more; not because there is more LGS around, but there is now a bigger rationale for labelling it [i.e., licensed treatments].”
Discussing the presentation of LGS in adults, Keough observed: “It tends to be a narrower spectrum of seizure types in adulthood than in younger children. In particular, the tonic nocturnal seizures take over as the primary seizure type, and seizures during wakefulness often become less prominent. Also, atypical absences are less prevalent in adults. Overall, the seizures become more subtle, and the seizure burden is lower. Patients tend not to injure themselves as much; although they may recede into a state of incapacitation, lose mobility, and become a lot more dependent, which can make their seizures seem less disruptive.”
Thomas added: “The brain is the last part of the body that fully matures, in the mid-20s, so often there are no more surprises in the adult clinic. The evolution has happened. If you have got an excellent paediatrician, then you will also get a lot of good information from the transition process in terms of drugs that work, drugs that have aggravated, drugs that are essential, and emergency strategies. Similarly, parents and carers will tell you about doctors who did not listen to them, or situations they do not want to repeat. So, there is a lot of information at this [adult] stage, and added to brain maturation, it means that often the situation is less volatile and more stable than in childhood. This makes it slightly easier to spot patterns, so it is a good time for initiating any changes in medicine as you are able to see whether it makes a difference.”
Amongst the main issues for diagnosis of LGS in adults, the experts highlighted an incomplete medical history, and lack of EEG evidence. “The challenges are evidence quality and depth,” said Thomas, “because you would always like to know more about what happened during that critical time of development [in childhood]. If an EEG was never done, never reported, or never kept, it is quite difficult to replicate the test. Then there is information synthesis, piecing it all together, and there are often red herrings too: the older you get, the more medical issues there are.”
Reinforcing this point, Keough added: “Due to many scenarios with older patients, there may be no one there to tell you what the child did when they were younger, and that makes it tricky because it is well known that the seizure mixture tends to simplify in older patients, and the EEG features might be absent too. It makes it harder to meet the diagnostic criteria.”
OTHER DIAGNOSTIC ISSUES
Detection of mild LGS, and identification of cases at the boundaries of the LGS definition, were other challenging aspects of diagnosis that were highlighted during the interviews. Keough commented: “People get a concept of LGS as being a refractory epilepsy that is devastating, but ‘devastating’ has levels of severity. There are some patients with mild intellectual disability and seizures that are less frequent, appearing less severe, who might be missed.”
Thomas agreed: “Disguised is a good word to use. Those with a mild learning disability might be advocating for themselves, and moving towards independent supported living, and with better, earlier treatments we might expect more people like that in the adult clinics.”
Keough remarked that the challenge is to “draw the circle broadly enough” to recognise all these cases, and Thomas commented: “It is sometimes quite easy to identify the patients at the ‘centre of the bullseye’ as they have got all the features, but working at the boundaries is always the challenge. I think it would be useful for prescribers to get together to discuss their thoughts on boundary cases and the diagnostic edge. The boundaries help you to tease out whether a case is or is not LGS, so it is more than [intellectual debate], it is the entry criterion for medication.”
TACKLING CHALLENGES IN DIAGNOSIS
Having discussed the challenges, the experts shared their thoughts on future improvements for the diagnosis of LGS, involving greater awareness and vigilance. “Within the epilepsy community, LGS is a really helpful term, but if you are outside the epilepsy world it is fraught with confusion. People do not always fully understand what LGS is,” said Thomas.
Keough agreed: “There is a huge lack of recognition of patients with LGS amongst adult neurologists; and for child neurologists who do not have epilepsy as their main focus, or have not had additional training, LGS seems like a very rare idea. Patients are put in a category of refractory epilepsy, with no thought beyond that to get to a syndromic diagnosis. So, now there is a lot of emphasis in neurologic training to try and raise awareness in healthcare providers to identify syndromes and make those diagnoses. In addition, in the USA we have been forced to be more vigilant about identifying the diagnosis in order to access treatments. You need to go in thinking about the diagnosis from the start. ‘Does this patient have LGS?’ ‘Do they meet all of the requirements?’ And again, you have to draw the circle broadly and widely, because it is such a heterogenous syndrome.”
Thomas also suggested that greater knowledge of the condition, its symptoms, and aetiology, could help drive improvements in diagnosis in the longer term. “I think an aetiology study would be very helpful at scale; a study of major centres, with a thousand people with LGS. It would also be interesting to know whether you could subdivide LGS into syndromes, because there is a variable level of learning deficit and some people, amazingly, become very nearly seizure-free in later life.” Thomas added: “I think EEG is important here as well. As we get used to having more long-term wearable EEG devices, we are going to get new understandings of disease evolution, as well as what the plateau period looks like in adults. The other thing that will happen is that we will have decision support tools. The first ones will be for epilepsy surgery, but it would not be inconceivable to have these tools to aid in the diagnosis of some syndromes too [linked to the ILAE criteria], and I think that would be a great support to physicians.
CONCLUSIONS
In these interviews, Keough and Thomas described LGS as a severe and refractory condition that is incompletely recognised. Diagnosis of LGS is key to managing expectations around disease course, accessing licensed treatments, and optimising seizure control. However, the experts highlighted many significant challenges around the diagnosis of LGS, including its typical early childhood onset, heterogeneous aetiology, and varied and constantly evolving presentation. In particular, they discussed the difficulties of diagnosing LGS in adulthood, when the seizure profile may be clouded by disease evolution and an incomplete medical history. Within this context, Keough and Thomas stressed the importance of the recently published ILAE diagnostic criteria for childhood epilepsy syndromes as a valuable and highly practical guide for identifying LGS in children (with some caveats around securing EEG criteria in adults). They also explained how greater physician awareness of the broad range of potential features of LGS, coupled with increased vigilance, could help to optimise care and quality of life for patients and families living with this severe epilepsy syndrome.






