Abstract
Colorectal cancer (CRC) is the most common gastrointestinal malignancy worldwide and one of the leading causes of cancer-related death. The overall management of CRC in different patient populations is complex and confounded by demographic, genetic, and lifestyle factors. Recent advances in early detection screening and treatment options have reduced CRC mortality in developed nations, even in the face of growing incidence. Biomarkers are an important component of CRC diagnosis, management, and prognosis. BRAF mutation is considered an important prognostic factor, along with other biomarkers, particularly in advanced stages of the disease. Detection of the BRAF V600E mutation in metastatic CRC identifies a subgroup of patients who derive little benefit from standard therapies and have an extremely poor prognosis. Recent data show that the addition of an anti-epidermal growth factor receptor agent to a BRAF inhibitor, with or without a MEK inhibitor, improves outcomes such as overall survival and response rates compared to irinotecan-based chemotherapy in patients with BRAF V600E-mutant CRC. As KRAS and NRAS mutation status is crucial for guiding therapy (e.g., prediction of lack of favourable effects with targeted anti-epidermal growth factor receptor therapies), all patients with metastatic CRC should undergo mutational testing. In addition, microsatellite instability status can predict possible advantages from immunotherapy and may also be applied to inform treatment decisions. With an evolving understanding of the pathophysiology of the disease and the pharmacology of the targeted drugs used in treatment, an individual approach to manage the disease seems to be the future of care.
INTRODUCTION
Colorectal cancer (CRC) is the most common gastrointestinal malignancy. Worldwide, it is the second most common cancer in females (614,000 cases, 9.2% of the total) and the third most common cancer in males (746,000 cases, 10.0% of the total). CRC is the fourth most common global cause of cancer-related mortality and the burden of this disease is expected to increase by 60.0% to >2.2 million new cases per year, and 1.1 million cases of cancer mortality per year by 2030.1 Trends in the incidence and mortality of CRC correlate with the Human Development Index (HDI) and a Western lifestyle; obesity, sedentary lifestyle, red meat and alcohol consumption, and tobacco smoking are considered important factors in CRC, with a high incidence of disease reported in Central and Eastern Europe and a low incidence reported in Africa.2,3 The overall management of CRC in different patient populations is complex and confounded by demographic, genetic, and lifestyle factors. Recent advances in early detection screening and treatment options have reduced CRC mortality in developed nations, even in the face of growing incidence.2
Approximately 50% of all patients with CRC will develop metastases and in 25% of the patients CRC is already metastasised at initial diagnosis.4,5 According to the European Society for Medical Oncology (ESMO) 2016 guidelines, if a physician has a clinical or biological suspicion that a patient may have metastatic CRC (mCRC), a biomarker analysis is required on the primary tumour or on the metastases at the time of diagnosis.6
Fluoropyrimidine-based chemotherapy was the backbone of CRC treatment, but the addition of irinotecan and oxaliplatin to form combination regimens has significantly improved overall survival.7 The 5-year survival for mCRC was approximately 60% in 2014.4 In the past decade, the development of novel biological agents, including therapies directed against vascular endothelial growth factor and epidermal growth factor receptor (EGFR), has further altered the landscape of mCRC treatment.7 Optimisation of systemic anticancer therapies, including new chemotherapeutic agents, immunotherapy, and targeted therapies, and early detection have greatly improved the prognosis of mCRC.4 Clinical studies indicate that some patients may respond differently to these treatments; therefore, individual patient- and tumour-related factors must be considered. A more tailored and biomarker-driven approach to treatment selection can optimise outcomes and avoid unnecessary adverse effects.7
SUBTYPES OF COLORECTAL CANCER
RAS and BRAF are downstream components of the EGFR signalling pathway. Knowledge of mutations in RAS and BRAF is required for frontline treatment decision-making for mCRC.8
Incidence of RAS Mutations
The RAS/RAF/MEK/ERK signalling cascade, also referred to as the MAPK pathway, is known to drive cell proliferation, differentiation, survival, and angiogenesis.9 EGFR helps regulate cell differentiation and survival via signalling upstream of the MAPK pathway.10 Dysregulation of this signalling pathway has been implicated as a critical mediator of tumourigenesis.9
RAS mutations were identified in 9–30% of all cancers (50% in CRC), with KRAS mutations the most dominant (86%), followed by NRAS (11%) and HRAS (3%).11-13
The RAS family of proto-oncogenes comprises HRAS, NRAS, and KRAS. RAS mutations are identified in approximately one-half of patients with mCRC.14 RAS mutations are used as predictive biomarkers for resistance to anti-EGFR therapies.15 Mutations in KRAS/NRAS exons 2 (codons 12 and 13), 3 (codons 59 and 61), and 4 (codons 117 and 146) are predictive of the lack of benefit of anti-EGFR therapy.8 Mutations in codons 12 or 13 of KRAS occur in approximately 40% of all mCRC and generate constitutive activation of the MAPK pathway. Data have shown that mutations in codons 61 and 146 of KRAS, or in codons 12, 13, and 61 of NRAS, occur in approximately 18% of patients with mCRC and can induce malignant transformation of colorectal cells in vitro.16-19 Previously classified as KRAS ‘wild-type’ tumours based on practices testing only for mutations in codons 12 or 13 of KRAS, these less common atypical RAS mutations have been shown to negate the presumed benefit of anti-EGFR therapies for patients with RAS wild-type tumours.16,17,19
BRAF-Mutant Colorectal Cancer
BRAF is a protein in the EGFR-mediated MAPK pathway; its downstream signalling activates MEK through its phosphorylation.20,21 BRAF mutations are found in 8–12% of cases of mCRC, with the predominance of BRAF V600E in approximately 90% of BRAF-mutant CRC.22-27 BRAF V600E is a point mutation at nucleotide 1799 that results in independent activation of its upstream activator protein, RAS, as well as increased stimulation of its downstream effector proteins, MEK and ERK, via phosphorylation.28 RAS and BRAF mutations are usually mutually exclusive.
BRAF-mutant CRC are associated with peculiar molecular, pathological, and symptomatic characteristics. BRAF V600E-mutant CRC occurs predominantly in females and in patients of older age, is often right-sided, mostly located in the proximal colon, presents with large tumour size with serrated components, exhibits peritumoural lymphoid reactions, and has poorly differentiated histology.29,30 BRAF V600E-mutant tumours often demonstrate frequent and multiple metastatic sites and increased rate of metastasis in the peritoneum and distant lymph nodes; however, lung localisations are relatively infrequent.18,31-33
It has been suggested that BRAF V600E CRC could be separated into two distinct subclasses based on gene expression: BRAF V600E mutant 1 (BM1) and BRAF V600E mutant 2 (BM2).34 BM1 is characterised by KRAS/AKT pathway activation, mTOR/4EBP deregulation, and epithelial-to-mesenchymal transition; BM2 is associated with vital deregulation of the cell cycle.30 Proteomics data validated these observations as BM1 is characterised by high phosphorylation levels of AKT and 4EBP1, and BM2 patients display high CDK1 and low cyclin D1 levels.30
The role of BRAF mutations (particularly V600E) in CRC and disease prognosis is gaining considerable research interest and there are substantial clinical data on the role of BRAF mutations and their impact on treatment decisions and prognosis. The application of RAS and BRAF genes as prognostic and predictive biomarkers has paved the way toward personalised treatment and management of patients with CRC.35 Biomarkers are useful to determine treatment algorithms in metastatic disease and enhance the use of proper systemic agents while planning patient management (Table 1).6,36-38
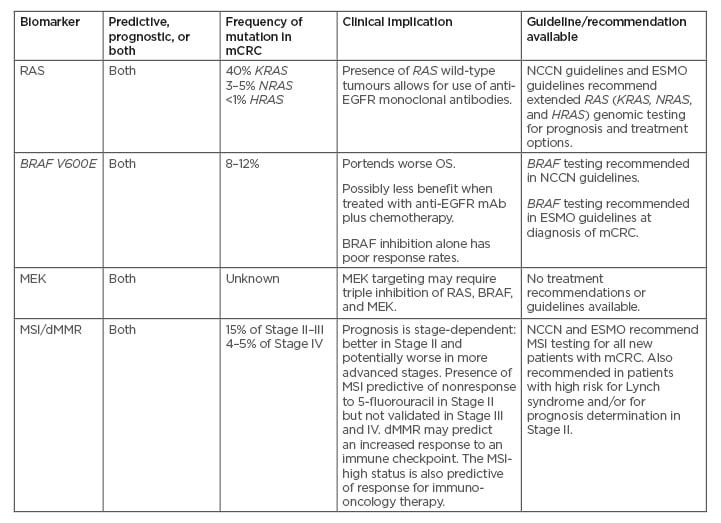
Table 1: Biomarkers and clinical implications in colorectal cancer.6,37,38
dMMR: deficient mismatch repair; EGFR: epidermal growth factor receptor; ESMO: European Society for Medical Oncology; mAB: monoclonal antibodies; mCRC: metastatic colorectal cancer; MSI: microsatellite instability; NCCN: National Comprehensive Cancer Network; OS: overall survival; WT: wild-type.
Evaluating Mutations at Diagnosis
At the time of metastatic disease diagnosis, patients should be evaluated for RAS mutation to enable strategic treatment decisions; also, tumour BRAF mutation should be evaluated for prognostic and predictive assessment. All patients considered for systemic therapy should be stratified according to whether their tumours are RAS-wild-type, RAS-mutant, or BRAF-mutant. A small subset of BRAF V600E tumours (up to 30%) demonstrate microsatellite instability (MSI).18 MSI testing has strong predictive value for the use of immune checkpoint inhibitors in the treatment of patients with mCRC and can also assist clinicians in genetic counselling.6 The evidence is growing that determining BRAF as well as MSI status at the time of diagnosis of metastatic disease is needed. Choosing the optimal treatment strategy for a patient requires accurate diagnosis and assessment of their disease.
TREATMENT OVERVIEW
Outcomes in BRAF-Mutant Tumours Versus BRAF-Wild-Type Tumours
The BRAF V600E mutation is an important prognostic and predictive factor for CRC, particularly in advanced stages of the disease.39 BRAF V600E mutations are associated with increased mortality risk, independent of age, sex, tumour location, MSI status, differentiation grade, and TNM grading. The BRAF V600E subtype is more aggressive (median overall survival [OS] for patients with BRAF-mutant V600E [BRAF-MT] mCRC: 11.4–18.2 months) than BRAF non-V600E (median OS: 60.7 months).18,40 The wild-type BRAF (BRAF-WT, no mutation) has a median OS of 41.1–43.0 months.18 Similar results were observed in a pooled analysis of four Phase III studies in patients with mCRC: patients with BRAF-MT had a worse prognosis compared to patients with BRAF-WT tumours (progression-free survival [PFS]: 6.2 versus 7.7 months; p<0.001; OS: 11.4 versus 17.2 months; p<0.001).41
Seligmann et al.42 evaluated data from 2,530 patients with advanced CRC who received chemotherapy in three randomised trials. A total of 231 patients (9.1%) were positive for BRAF mutation, although the clinicians were previously unaware of this. Patients with BRAF-WT tumours and BRAF-mutant tumours administered first-line oxaliplatin/fluorouracil had comparable PFS (5.7 versus 6.3 months; p=0.26) and disease control rate (59.2% versus 72%; p=0.24). Patients with BRAF-mutant tumours had a significantly shorter post-progression survival (4.2 versus 9.2 months; p<0.001) after advancement on first-line chemotherapy.36 The unfavourable prognosis of patients with BRAF V600E tumours may be because of resistance to chemotherapy after first-line treatment. In these patients, response to standard chemotherapy is limited and therefore the response rate is poor.43
Conversely, the benefit of immunotherapy agents in MSI deficient mismatch repair (dMMR) tumours does not appear to be significantly affected by BRAF mutational status. For example, in CheckMate 142, an open-label, multicentre, Phase II study, nivolumab alone or in combination with ipilimumab was associated with meaningful benefit (objective response rate [ORR]: 25%; disease control rate [DCR]: 75%; and ORR: 55%; DCR 80%, respectively) in patients with previously treated mCRC with MSI-high BRAF-MT.44,45
Patients with BRAF-MT more commonly develop peritoneal metastases, less frequently present with disease limited to the liver, and have shorter survival after metastasectomy compared to patients with BRAF-WT tumours.46 Although these data indicate that patients with BRAF-MT are less likely to benefit from metastasectomy compared to those with BRAF-WT tumours, Margonis et al.47 argue that current evidence cannot support denying metastasectomy in patients with otherwise resectable disease solely based on their overall KRAS or BRAF mutational status.
Microsatellite Instability and Related Treatments and Outcomes
The MSI phenotype results from dMMR because of germline mutations in MMR genes or from epigenetic silencing of the MLH1 gene, with mutations in the BRAF oncogene in the majority of cases.48 The MSI phenotype occurs in 10–20% of all patients with CRC and is more common in Stage II disease (approximately 20%) than in Stage III (approximately 12%) and metastatic disease (4%).49 BRAF V600E mutations were reported in 30% of MSI tumours, whereas MSI tumours were reported in 29% of BRAF V600E-positive mCRC.18 The prognosis and outcome in patients with mCRC and MSI-high are often poor. MSI tumours, like BRAF-mutated CRC, have specific clinicopathological features, such as primary location on the right side, poor differentiation, mucinous histology, and increased numbers of tumour-infiltrating lymphocytes.50,51 BRAF V600E mutation is not a reliable prognostic factor in MSI-high mCRC.52
The relationship between BRAF mutation and MSI in terms of diagnostic and prognostic implications is complex. In a pooled analysis of four Phase III studies (CAIRO, CAIRO2, COIN, and FOCUS) in first-line treatment of mCRC, median PFS and OS were significantly poorer for patients with dMMR compared to proficient MMR tumours (hazard ratio [HR]: 1.33; 95% confidence interval [CI]: 1.12–1.57; and HR: 1.35; 95% CI: 1.13–1.61, respectively) and for patients with BRAF-MT compared to BRAF-WT tumours (HR: 1.34; 95% CI: 1.17–1.54; and HR: 1.91; 95% CI: 1.66–2.19, respectively).35 PFS and OS were significantly decreased for patients with BRAF-MT within the group of patients with proficient MMR, but not for BRAF status within dMMR, or MMR status within BRAF-WT or BRAF-MT.46 These results indicate that the poor prognosis of MSI metastatic tumours appears to be largely driven by the coexisting BRAF mutation.
MSI-high has been associated with favourable survival in early-stage BRAF V600E-mutant mCRC; however, the BRAF V600E mutation has been associated with a poorer survival rate in microsatellite stable mCRC.53,54 The effect of BRAF and KRAS mutations on survival was assessed in patients with Stage II and III MSI colon cancer.45 Patients with double wild-type cancers (i.e., BRAF and KRAS wild-type) had a highly favourable survival with 5-year cancer-specific survival of 93% (95% CI: 84–100%), whereas patients with cancers harbouring mutations in either BRAF or KRAS had 5-year cancer-specific survival of 76% (95% CI: 67–85%). On multivariate analysis, mutation in either BRAF or KRAS versus double wild type remained significantly prognostic.45
TREATMENTS
Treatment options for mCRC depend on the stage of disease, tumour location, performance status, comorbidity, organ function of the patient, and the molecular makeup of the tumour. Every therapeutic choice must integrate patient-related factors and preferences; tumour features, such as molecular profile and burden of disease; treatment goals; and accessibility. Therapeutic goals in the management of patients with mCRC can be OS, PFS, symptom improvement, and/or maintaining quality of life. The first line of treatment is important as it determines the subsequent lines of therapy and thus the continuum of care; however, the therapeutic goals will change according to the line of treatment administered. For example, the goal of tumour size reduction is more relevant in first-line treatment, whereas disease control and palliation of tumour-related symptoms is more relevant in subsequent lines of treatment.55 Lines of treatment are particularly important when considering patients with BRAF-mutant mCRC as not many of these patients receive second- or third-line treatment. In an analysis of outcomes of patients with mCRC in randomised clinical trials, fewer patients with BRAF-mutant mCRC received second-line treatment compared to patients with BRAF-WT mCRC (33% versus 51%; p<0.001).36 In a further study of a prospective population-based mCRC cohort, patients with BRAF-mutant mCRC received first- and second-line chemotherapy just as often as patients with BRAF-WT mCRC (first-line: 52 [57%] versus 225 [64%]; second-line: 28 [30%] versus 132 [37%]), but fewer received third- and fourth-line chemotherapy (third-line: 5 [5%] versus 66 [19%]; fourth-line: 1 [1%] versus 26 [7%], respectively).56
For patients with a BRAF V600E mutation, recent combination strategies targeting BRAF, EGFR, and MEK have been more successful, with improved response rates (Table 2).57-70 A more tailored and biomarker-driven, personalised approach to treatment selection can optimise outcomes and avoid unnecessary adverse effects of cytotoxic treatment combinations.7 The expanded use of biomarkers to guide the treatment of patients with CRC has revealed a level of complexity arising from interactions between different biomarkers. An improved understanding of the causes of primary resistance may increase response rates among patients receiving targeted therapies and enable more effective drug combinations, exemplified by mutations in the MAPK signalling pathway for EGFR-targeted and/or BRAF-targeted therapies.71 This review summarises the current landscape and emerging data on the molecular, clinical, and therapeutic aspects of BRAF V600E CRC.
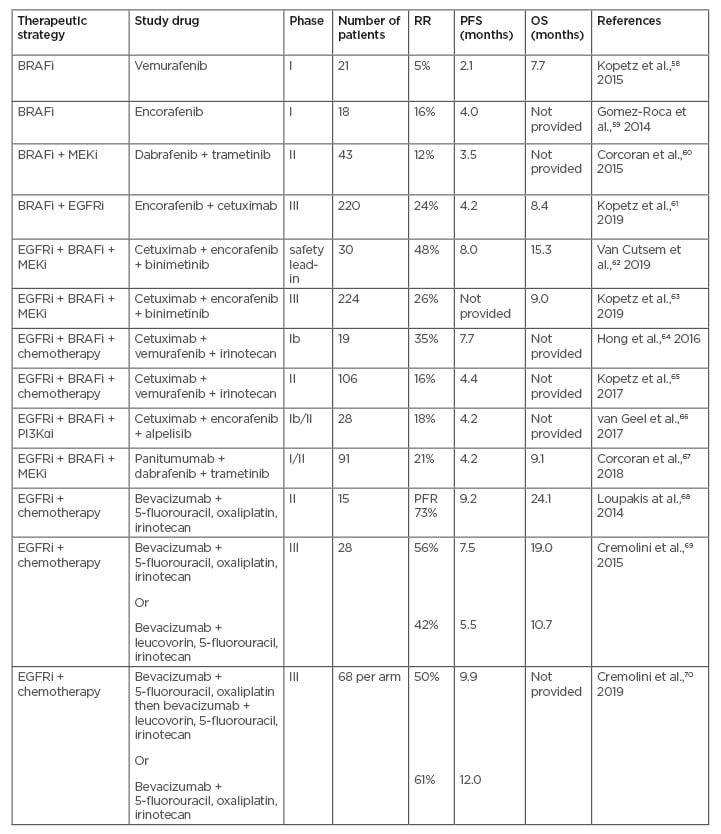
Table 2: Overview of clinical trials investigating combination targeted treatments in patients with BRAF-mutant metastatic colorectal cancer.
BRAFi: BRAF inhibitor; EGFR: epidermal growth factor receptor; MEKi: MEK inhibitor; OS: overall survival; PFR: progression-free rate; PFS: progression-free survival; PI3Kαi: phosphoinositide 3-kinase α inhibitor; RR: response rate.
Adapted from Lee HM et al.,57 2019.
Intensive Chemotherapy
Rigorous chemotherapy combining 5-fluorouracil, oxaliplatin, and irinotecan (FOLFOXIRI) and the anti-vascular endothelial growth factor-A antibody bevacizumab may provide satisfactory outcomes.1 A prospective Phase II trial assessed the PFS of 15 (7%) patients with BRAF-mutant mCRC tumours treated with first-line FOLFOXIRI plus bevacizumab.68 At a median follow-up of 25.7 months, the 6-months progression-free rate was 73%. Median PFS and OS were 9.2 and 24.1 months, respectively, highlighting that this combination therapy may be a reasonable option for first-line treatment of patients with BRAF-mutant mCRC.54 In the Phase III TRIBE study, the efficacy of FOLFOXIRI plus bevacizumab was compared to that of leucovorin, 5-fluorouracil, and irinotecan (FOLFIRI) plus bevacizumab.69 FOLFOXIRI plus bevacizumab was efficacious in the treatment of CRC irrespective of BRAF status. Furthermore, in the BRAF-mutant subgroup (28 patients), patients treated with FOLFOXIRI plus bevacizumab achieved a median PFS of 7.5 months versus 5.5 months in patients treated with FOLFIRI plus bevacizumab. Patients with BRAF-MT in the FOLFOXIRI plus bevacizumab group also achieved an 8-month improvement in median OS (19.0 months versus 10.7 months; HR: 0.54; 95% CI: 0.24–1.20); however, the difference was not statistically significant.55 In the Phase III TRIBE2 study, patients with unresectable mCRC were randomised 1:1 to first-line FOLFOX plus bevacizumab followed by FOLFIRI plus bevacizumab after disease progression (PD) (Arm A), or FOLFOXIRI plus bevacizumab followed by reintroduction of the same regimen after PD (Arm B).70 The upfront treatment of FOLFOXIRI plus bevacizumab followed by reintroduction of the same regimen after PD increased PFS2 (the time from randomisation to PD on any treatment given after first PD or death) (19.1 versus 17.5 months; HR: 0.74; 95% CI: 0.62–0.88; p<0.001), first PFS (12.0 versus 9.8 months; HR: 0.75; 95% CI: 0.63–0.88; p<0.001), and OS (27.6 versus 22.6 months; HR: 0.81; 95% CI: 0.67–0.98; p=0.033) compared to a preplanned sequential strategy of FOLFOX plus bevacizumab followed by FOLFIRI plus bevacizumab. Efficacy results reported were consistent with the results from the previous TRIBE study. Treatment effect was consistent across all analysed clinical and molecular subgroups in terms of both PFS2 and first-PFS, with potentially increased benefit among patients with a right-sided and/or a RAS– or BRAF-mutant tumour, and particularly among those with Eastern Cooperative Oncology Group (ECOG) performance score 0.56 According to the ESMO 2016 guidelines, the preferred first-line treatment for patients with BRAF-mutant CRC is FOLFOXIRI plus bevacizumab for cytoreduction (tumour shrinkage) and disease control (control of progression).6 The TRIBE2 data (68 patients with BRAF-mutant CRC per arm) showed that compared to FOLFOX plus bevacizumab, upfront FOLFOXIRI plus bevacizumab significantly improved PFS1 (median 12.0 versus 9.9 months; HR: 0.73; 95% CI: 0.62–0.87; p<0.001) and response evaluation criteria in solid tumours response rate (61% versus 50%; OR: 1.55; 95% CI: 1.14–2.10; p=0.005).56
BRAF Inhibitors (Vemurafenib, Dabrafenib, and Encorafenib)
Treatment with BRAF inhibitors causes a rapid feedback activation of EGFR that leads to proliferation of BRAF V600E-MT cells. Therefore, in patients with CRC with known BRAF mutation, single-agent BRAF inhibitors appear to be ineffective.72 This contrasts with treatment in other tumours with BRAF mutation such as melanomas, because melanoma cells express low levels of EGFR and are therefore not subject to this feedback activation. In mutation-positive patients, treatment with vemurafenib, dabrafenib, or encorafenib as single agents was inadequate; however, combining these agents with therapies targeting the MAPK pathway may overcome the primary resistance to BRAF inhibition and provide a good therapeutic option for these patients.21,73 Clinical trials combining BRAF (and MEK) inhibitors, either singly or in combination with the anti-EGFR antibodies cetuximab or panitumumab, showed improved efficacy; however, the response rates were heterogeneous.71 Other approaches that are being investigated in BRAF V600-mutant mCRC are BRAF inhibitors in combination with a PI3K inhibitor.66
BRAF inhibition leads to the upregulation of a variety of receptor tyrosine kinases in CRC cell lines, including EGFR, human epidermal growth factor receptor (HER)2, and HER3. Combination of the BRAF-inhibitors vemurafenib, dabrafenib, or encorafenib with inhibitors dually targeting the EGFR and HER2 (such as lapatinib, canertinib, and afatinib) significantly reduced the metabolic activity and proliferative potential of CRC cells.71 This resensitisation was also observed after genetic depletion of HER2 or HER3. Dual HER2-targeted therapy causes tumour regression, and HER2-mutated cell lines, but not KRAS-mutated cell lines, are sensitive to the tyrosine kinase inhibitors neratinib and afatinib, and provide strong preclinical rationale for clinical trials targeting HER2-activating mutations in mCRC.74
EGFR Inhibitors (Cetuximab and Panitumumab)
The role of BRAF mutation as a predictive biomarker for anti-EGFR monoclonal antibodies (mAb) is controversial and there is a paucity of robust data to address this issue, mainly because of the small proportion of patients with BRAF-MT in the clinical trials. In a meta-analysis by Rowland et al.75 that evaluated the effect of BRAF-MT on the treatment benefit from anti-EGFR mAb for mCRC, there was insufficient evidence to definitively state that individuals with RAS-WT/BRAF-MT attained a different treatment benefit from anti-EGFR mAb for mCRC compared to individuals with RAS-WT/BRAF-WT.75
As the BRAF protein is localised directly downstream of the RAS protein, therapies targeting anti-EGFR mAb gained interest for the management of patients with BRAF-MT CRC. A retrospective multicentre analysis of the effect of downstream mutations on the efficacy of cetuximab showed that patients with BRAF-MT had a significantly lower response rate (8.3% versus 38.0%; p=0.0012) and DCR (37.5% versus 77.3%; p<0.0001) compared to patients with BRAF-WT.13 Median PFS (8 weeks versus 26 weeks; p<0.0001) and OS (26 weeks versus 54 weeks; p<0.0001) were also shorter in the patients with BRAF-MT.13 Other studies have also not confirmed the efficacy of anti-EGFR agents alone or with chemotherapy in patients with BRAF-MT.16,76,77 However, it is increasingly accepted that the addition of an EGFR inhibitor to a cytotoxic combination does not increase the activity of the cytotoxic regimen in patients with a BRAF V600E mutation.6
Targeting BRAF and EGFR
Hyman et al.78 studied 122 patients with BRAF V600E mutation-positive cancer, of which 37 had CRC and received vemurafenib with (27 patients) or without (10 patients) cetuximab.78 In the subgroup of patients with mCRC who received vemurafenib monotherapy, median PFS was 4.5 months (95% CI: 1.0–5.5) and median OS was 9.3 months (95% CI: 5.6–not reached). In patients who received combination therapy of vemurafenib and cetuximab, median PFS was 3.7 months (95% CI: 1.8–5.1) and median OS was 7.1 months (95% CI: 4.4–not reached). In the cohort of patients with CRC who received combination therapy, approximately 50% of patients had tumour regression that did not achieve partial response. Another study that evaluated the efficacy and safety of panitumumab and vemurafenib in 15 patients with BRAF-mutant mCRC reported 10 tumour regressions in 12 evaluable patients; two patients had a partial response and two had stable disease lasting over 6 months with acceptable tolerability.79
Rationale of Inhibitor Combinations
Clinical research has shown that targeting a single mutated gene or biomarker in CRC does not yield the desired clinical benefit. Meanwhile, preclinical research has provided more insight into the interactions between various signalling pathways and has shown that single kinase protein inhibition is unaffected by the feedback escape mechanisms and dynamic interactions between pathways. The road to clinical success of combining targeted therapies may be impacted by the requirement to show efficacy and predict drug responses. Hence, it is important to investigate the utility of cancer models, such as patient-derived mouse xenograft models for cancer and three-dimensional cell culture systems for predicting drug responses.80 Combined inhibition showed a strong synergistic effect in vitro and in xenograft models and resulted in complete inhibition of tumour growth.81 Furthermore, the addition of a MEK inhibitor to BRAF and EGFR inhibitors increased inhibition of ERK signalling and targeted both potential mechanisms of resistance to BRAF inhibitors. A Phase II study evaluated the combination of dabrafenib, panitumumab, and trametinib in patients with BRAF V600E-mutant mCRC.67 The response rate for the triplet therapy was 21% (95% CI: 13.1%–30.7%) compared to 10% in the dabrafenib plus panitumumab arm (95% CI: 1.2%–31.7%). These results indicate a role for combined targeted therapies as a therapeutic option for BRAF-mutant disease.78 Adverse events of Grade III or higher occurred in 70% of patients in the triplet-therapy group, 45% in the dabrafenib plus panitumumab group, and 67% in the trametinib plus panitumumab group.78 The most common adverse events of Grade III or higher were rash, dermatitis acneiform, diarrhoea, and fatigue in the triplet-therapy group; dry skin, hypomagnesaemia, and constipation in the dabrafenib plus panitumumab group; and dermatitis acneiform, dry skin, and rash in the trametinib plus panitumumab group. In another Phase II study (SWOG S1406), patients with BRAF V600E-mutated mCRC were randomised to irinotecan and cetuximab with or without vemurafenib.65 Median PFS without vemurafenib was 2.0 months and with vemurafenib was 4.4 months (HR: 0.42; 95% CI: 0.26-0.66; p<0.001). Similarly, response rate was 4% versus 16% (p=0.08), and DCR was 22% versus 67% (p=0.001).78 The Phase III BEACON CRC study randomised 665 patients with BRAF V600E-mutant CRC who had progressed after one or two prior regimens in the metastatic setting to either encorafenib plus cetuximab (doublet therapy); encorafenib, cetuximab, and binimetinib (triplet therapy); or the investigator’s choice of irinotecan or leucovorin, fluorouracil, and irinotecan (FOLFIRI) and cetuximab.62,65,67 Median OS was 9.0 months (95% CI: 8.0–11.4) in the triplet targeted-therapy arm compared to 5.4 months (95% CI: 4.8–6.6) for control (HR: 0.52; 95% CI: 0.39–0.70; p<0.001).67 Median OS in the doublet-therapy group was 8.4 months (95% CI: 7.5–11.0), and the risk of death was significantly lower than in the control group (HR: 0.60; 95% CI: 0.45–0.79; p<0.001).67 Updated OS data for this study (6 months of additional follow-up) were presented at the 2020 American Society of Clinical Oncology Gastrointestinal Cancers (ASCO GI) Symposium: median OS was 9.3 months in the triplet-therapy group, 9.3 months in the doublet-therapy group, and 5.9 months in the control group.82 Confirmed ORR by blinded central review for the triplet-targeted therapy was 26% (95% CI: 18–35) compared to 2% (95% CI: 0–7; p<0.001) for the control group.67 ORR in the doublet-therapy group was 20% (95% CI: 13–29), which was also significantly higher than that of the control group (p<0.001).67 Therefore, encorafenib, cetuximab, and binimetinib (triplet therapy), and encorafenib plus cetuximab (doublet therapy) significantly improved OS and ORR compared to the current standard of care (control) in pretreated patients with BRAF V600E-MT mCRC. The safety and tolerability profiles of both combinations were consistent with the known profiles of the individual component agents. Adverse events of Grade III or higher occurred in 58% of patients in the triplet-therapy group, 50% in the doublet-therapy group, and 61% in the control group.67 The most common adverse events of Grade III or higher were diarrhoea, abdominal pain, and nausea in the triplet-therapy group; fatigue, asthenia, and abdominal pain in the doublet-therapy group; and diarrhoea, asthenia, and abdominal pain in the control group. The trial was not powered to compare the two experimental groups (triplet combination therapy and doublet therapy) directly. The results from this prospective, randomised, Phase III study are the first evidence of survival benefit from biomarker-defined patients with mCRC who were administered a chemotherapy-free targeted regimen, thus defining a new standard of care. There are other targeted agents available, such as bevacizumab and regorafenib, but these are not specifically for patients with BRAF-mutant mCRC.
SURVEILLANCE
Younger patients with CRC often receive more aggressive treatment regimens than older patients, without a corresponding improvement in survival.83 In elderly patients with CRC, comorbidity and frailty are strong prognostic factors of survival, in addition to the commonly considered sociodemographic and tumour characteristics.84 Comprehensive geriatric assessment and early identification and management of comorbidities and geriatric syndromes are necessary to optimise care in elderly patients with CRC. Life expectancy for noncuratively managed patients with CRC is low, and some patients require subsequent surgical intervention for palliation; in a retrospective audit by Thavanesan et al.,85 life expectancy was 6.8 months, with one in nine patients requiring surgery for palliation.
The poor prognosis of patients with BRAF V600E tumours may be secondary to rapid progression following first-line treatment and reduced use of subsequent lines of therapy.36,39 To address this, close surveillance strategies need to be implemented in routine clinical practice to ensure the prompt initiation of second- or further-line treatment in such patients.
ONGOING AND PLANNED TRIALS
Research continues in other treatment combinations in BRAF-mutant mCRC.5 Binimetinib is being used in several ongoing trials with various combination regimens, including immunotherapy and chemotherapy.5 Results from preclinical and clinical trials indicate that a BRAF inhibitor and/or a MEK inhibitor might have immunomodulatory effects, leading to increased infiltration of immune cells into the tumour and a better functionality of immune effector cells.5,86-88 A better understanding of pharmacokinetics, pharmacodynamics, and pharmacogenetics is required to optimise synergy of BRAF inhibitors and/or MEK inhibitors and immunotherapy.5,89 Binimetinib combined with pembrolizumab and bevacizumab is currently being assessed in a Phase II trial in patients with refractory mCRC, including those with BRAF-MT disease.90 Recruitment is completed in a Phase I/II trial in which binimetinib is combined with immunotherapy in KRAS-mutant, microsatellite-stable mCRC;91 the results of this trial may expand indications for the use and combination of binimetinib.5 Patients are currently being recruited into a Phase I/II trial of encorafenib and binimetinib combination in advanced non-V600 activated BRAF-mutant cancers.92 A trial investigating encorafenib, binimetinib, and cetuximab in a first-line setting is ongoing to explore the efficacy in treatment-naïve patients with mCRC.5,93 Further trials are being conducted with dabrafenib in combination with other targeted agents and immunotherapy.94-96
CONCLUSION
BRAF V600E mutation in mCRC identifies a subgroup of patients who derive little benefit from standard treatments and have an extremely poor prognosis. Response rates in patients with BRAF V600E-MT CRC are lower compared to other BRAF V600E-MT advanced cancers when treated with BRAF-inhibitor monotherapy. The addition of targeted therapies against EGFR and/or MEK to BRAF inhibitors improves outcomes for patients with BRAF V600E-MT mCRC.
There is a need to increase the weight of data for patients with BRAF-MT mCRC as clinical studies conducted so far include only low numbers of these patients. This is particularly important considering the increasingly heterogeneous nature of this disease, including within the BRAF-MT group, in which patients with BRAF non-V600E-MT mCRC fare better than patients with BRAF V600E-MT mCRC.
Mutation testing for any patient with mCRC will help to guide management and treatment of the disease. Testing at diagnosis in line with the ESMO guidelines is key for optimising the treatment strategies for these patients.
The field continues to evolve in terms of new treatment strategies, which is offering some hope for patients with poor prognosis. The positive data from the BEACON CRC study and results with immunotherapy in patients with MSI-high BRAF V600E-MT mCRC are encouraging for future treatment of these patients.
The monitoring of these patients is also evolving, with circulating tumour DNA (ctDNA) providing an early indication of tumour response or tumour progression, which will enable an earlier switch to different treatments.97,98 There are considerable data correlating ctDNA and PD, thus providing a potential method to screen these patients more effectively. As these patients progress so rapidly, it is problematic to wait 6–8 weeks to conduct a scan to check progress; ctDNA could help circumvent this issue and may become standard practice in the future.
Further clinical trials to investigate different patient subsets and to assess the effects of earlier lines of treatment will also add to the clinical picture for BRAF V600E-MT mCRC.






