Meeting Summary
Taking place during the 2025 European Respiratory Society (ERS) International Congress held in Amsterdam, the Netherlands, this symposium presented up-to-date expert perspectives on the challenges of advanced lung disease, focusing on alpha 1 antitrypsin deficiency (AATD) and bronchiectasis.
Experts discussed the latest findings regarding the real-world management of AATD, including how to assess lung and liver function in patients, when and how to initiate augmentation therapy, and the importance of exercise and pulmonary rehabilitation. The current understanding of bronchiectasis was discussed, and experts shared their thoughts on differentiating disease severity from disease activity, and the value of precision medicine in managing the disease.
Key messages from the symposium included the importance of recognising that AATD can manifest with extra-pulmonary disease, the growing evidence supporting a survival benefit of augmentation therapy in AATD, and the rapid advances in the field of bronchiectasis that are revealing new treatment targets and opportunities for precision medicine.
Perspectives on Alpha 1 Antitrypsin Deficiency
One of the most common genetic diseases, AATD, involves the retention of misfolded alpha 1 antitrypsin (AAT) in hepatocytes, which can cause liver cirrhosis. In case of an inflammatory insult, the consequent deficiency of AAT in the circulation results in unopposed protease activity, perpetuating inflammation and leading to progressive tissue damage, including emphysema in the lungs and panniculitis in the skin (Figure 1).1,2
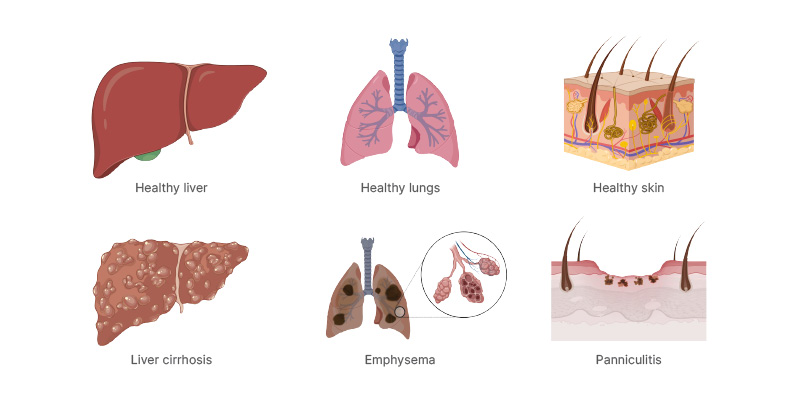
Figure 1: Sites of disease in alpha 1 antitrypsin deficiency.
Created with BioRender.com.
Adapted from McElvaney et al.2
The latest findings regarding the real-world management of AATD were discussed by Gerry McElvaney, Professor of Medicine, Royal College of Surgeons in Ireland (RCSI), University of Medicine and Health Sciences, Dublin, Ireland; and Emily F.A. van ‘t Wout, Assistant Professor and treating physician, Department of Pulmonology, Leiden University Medical Centre, the Netherlands.
Real-World Alpha 1 Antitrypsin Deficiency: What to Monitor, Measure, and Manage in the Clinic
Assessing lung function
McElvaney presented data from a 2024 Delphi consensus of 103 members of the European Alpha 1 Research Collaboration (EARCO) group, which focused on the optimal follow up of patients with AATD and advanced lung disease.3
In terms of assessing lung function in worsening disease (defined as one or more of moderate-severe dyspnoea, 1–2 exacerbations per year requiring hospitalisation, or forced expiratory volume in one second [FEV1] decline of ≥50 mL/year), the panel agreed that both pre- and post-bronchodilator spirometry measurements should be performed at baseline, whereas subsequent measurements could be limited to post-bronchodilator application.3 Spirometry parameters should include FEV1, forced vital capacity (FVC), the FEV1/FVC ratio, inspiratory vital capacity (VC), maximal VC, and inspiratory capacity. To assess lung decrement prior to changes in FEV1 and FVC, forced expiratory flow rates (FEF25, FEF50, and FEF75) as a percentage of FVC, and maximal expiratory flow rates (MEF75, MEF50 and MEF25) as a percentage of remaining VC, should also be measured. Body plethysmography parameters should include total lung capacity (TLC), residual volume, residual volume/TLC, airway resistance, specific airway resistance, functional residual capacity, and functional residual capacity/TLC. Chest X-ray should be performed only when clinically indicated, and arterial blood gas parameters (pH, partial pressure of O2, partial pressure of CO2, hydrogen carbonate ion) should be assessed via arterial puncture.
While the panel consensus was that FEV1 should be used to follow up patients with AATD, McElvaney stressed that changes in this parameter may be minimal among patients with lung disease at diagnosis (lung-index cases), since real-world evidence suggests that these patients experience a plateau in FEV1 decline from their mid-40s (Figure 2).4
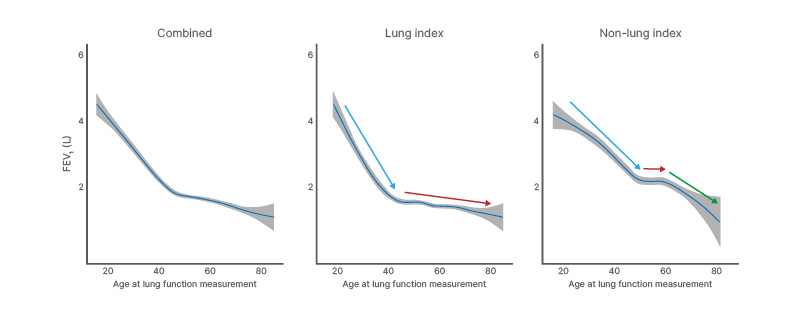
Figure 2: Forced expiratory volume in one second decline is non-linear, with an inflection point in the late 40s and different trajectories for lung-index and non-lung index patients.
Adapted from Fraughen et al.4
FEV1: forced expiratory volume in one second.
When and how should augmentation therapy be initiated?
The EARCO panel agreed that patients should have AAT levels <11 µM and CT-confirmed emphysema prior to initiation of AAT augmentation therapy, and that patients should abstain from smoking for >6 months.3 McElvaney also noted that AAT levels alone are not sufficient to make a diagnosis of AATD and should be accompanied by genotyping, phenotyping, and/or gene sequencing as indicated. The age of the patient and the deterioration of FEV1 should also be considered, and it was generally agreed that therapy should not be initiated following lung transplantation. During augmentation therapy, clinicians should consider an extended dosing interval (off-label; follow local guidelines), and AAT levels should be monitored at trough levels whenever there are changes in dose or treatment interval, or significant changes in the patient’s BMI.3
In a large observational study, augmentation therapy has been shown to confer a survival advantage in AATD,4 with a substantial, unadjusted life-years gain of 6.9 years versus standard care over a 50-year horizon.5 However, longitudinal lung function measurements identified distinct AATD phenotypes.4 In non-lung-index patients, augmentation therapy was not associated with a reduction in FEV1 decline, while in lung-index patients, the only group for whom therapy was associated with a significant reduction of decline was those with relatively well-preserved lung function.4 McElvaney emphasised that these data further highlight the potential difficulties of using FEV1 as a primary follow-up measure.
What about alpha 1 antitrypsin deficiency liver disease?
Liver disease associated with AATD can range from mild, self-limiting cholestasis in infancy to chronic hepatitis, cirrhosis, hepatocellular carcinoma (HCC), or even fulminant hepatic failure.6 Approximately 4% of children with AATD experience life-threatening liver disease,6,7 and McElvaney stressed that infants with severe liver disease may enter a ‘honeymoon period’ where they experience normal growth and few signs or symptoms of the disease, followed by renewed progressive injury and decompensation in their teenage years.6 McElvaney emphasised the need for early involvement of hepatologists in the care of people with AATD.
Risk factors for liver fibrosis in adults with protease inhibitor ZZ (PiZZ)-genotype AATD include age >50 years, male sex, obesity, diabetes, high BMI, and metabolic syndrome.8 A retrospective review found that 7.9% of explant livers from adults undergoing liver transplantation had periodic acid-Schiff globules suggestive of AATD, yet less than two-thirds of these patients were tested for AATD, indicating that diagnosis of AATD is often overlooked in this patient population.9
Vibration-controlled transient elastography is the preferred non-invasive test for liver fibrosis.10 While magnetic resonance elastography was considered highly sensitive and specific for detecting advanced fibrosis, this technique is limited by cost and lack of general availability.10 Traditional ultrasound can be useful in screening for HCC or liver cirrhosis; the detection of steatosis; the identification of nodular, structural, or biliary liver disease; and the monitoring of fibrosis progression in patients with BMI >35 kg/m2, but it is considered to have limited sensitivity for differentiating progression in early fibrosis.10 Recommendations regarding the frequency of liver monitoring depend on the degree of fibrosis. Mild fibrosis should be evaluated every 2–3 years, whereas moderate-to-severe fibrosis should be evaluated every 6–12 months.10
The transition from F1 (portal fibrosis without septa) to F2 (portal fibrosis with few septa) on the Meta-analysis of Histological Data in Viral Hepatitis (METAVIR) scale is considered to be the most important aspect of risk stratification for liver disease in AATD.10 Clinical signs of advanced liver disease (including portal hypertension, hepatic encephalopathy, and sarcopenia) or HCC may indicate that a patient with AATD should be considered for liver transplant.10
Does alpha 1 antitrypsin deficiency always present with lung or liver disease?
To emphasise that not all incidents of AATD present with one of these two common manifestations, McElvaney described the case of a 23-year-old male who initially presented to a tertiary referral centre with an acutely swollen right knee and joint effusion (mainly neutrophilic). The patient was discharged on non-steroidal anti-inflammatory medication; however, 8 weeks later, he presented to a different centre with chest pain and severe dyspnoea, and was found to have bilateral pleural effusions (low pH exudate, mainly neutrophilic). C-reactive protein levels remained very high after attempted drainage of the effusions, and he was admitted to the ICU with respiratory failure. At this stage, the patient recalled that he had had jaundice as an infant and had been told he had a rare disease. The medical team tested for AATD, which led to a diagnosis of acute serositis/panniculitis secondary to PiZZ AATD. Due to inflammation, the patient’s AAT levels were unusually high. Treatment with high-dose intravenous AAT was initiated (off-label), and the patient’s symptoms resolved within 12–24 hours. When the patient presented again, 6 weeks later, with a diffusely swollen, tender right upper limb, AAT augmentation therapy was reinstituted, and the patient remains on maintenance high-dose therapy.
The Nuts and Bolts of Caring for Patients with Alpha 1 Antitrypsin Deficiency
van ‘t Wout explained that AAT mutations can vary considerably, and in the most frequent phenotype, PiZZ, this results in both retention of AAT in the liver (leading to liver disease) and a deficiency in circulating AAT (predisposing individuals to early onset emphysema).1
However, van ‘t Wout emphasised that, while medical textbooks state that emphysema in AATD is typically panacinar and basal, in real life, ‘classical’ centrilobular emphysema of the upper lobes can also be found, especially with increasing age.1 Patients may also present with other pulmonary manifestations, such as bronchiectasis, asthma, or vasculitis associated with anti-neutrophil cytoplasmic antibody.1,11 Of note, in a retrospective cohort study in the USA, only 5.6% of patients with newly diagnosed COPD and liver disease were tested for AATD between 2012–2021,12 and the testing rates for patients with asthma and bronchiectasis are even lower.
Inhalation therapy
Inhalation therapy is commonly used to treat pulmonary symptoms of AATD, although no AATD-specific therapies are available. van ‘t Wout explained that clinicians generally follow COPD guidelines regarding the use of long-acting β-agonist or muscarinic antagonists to treat their patients with AATD.13-15 In cases with comorbid asthma, inhaled corticosteroids may also be used.14 This can increase FEV1 and exercise capacity, and decrease dyspnoea.16 Treatment may also depend on the phenotype of the AATD-associated lung disease, since frequent exacerbations are associated with a greater decline in lung function compared with infrequent exacerbations,17 and bacterial colonisation of the lower respiratory tract is associated with a substantially increased risk of exacerbations compared with no colonisation.18 van ‘t Wout cautioned clinicians to consider the impact of inhaler choice on climate change, and stressed that pressurised measured dose inhalers contain hydrofluorocarbons, which impact global warming.19
Alpha 1 antitrypsin augmentation therapy
AAT augmentation therapy in PiZZ AATD has been associated with a significant reduction in lung density loss, but with less impact on exacerbation frequency and quality of life (QoL).20,21 Findings vary in terms of the impact of augmentation therapy on FEV1 decline,22,23 with differences potentially depending on the AAT variant (van ‘t Wout, unpublished data). Real-world, long-term data show that augmentation therapy is associated with reduced mortality,4 but not with improvements in QoL.24
Exercise and pulmonary rehabilitation
High levels of sedentary behaviour are associated with poorer cardiometabolic health, whereas high levels of physical activity are associated with improved cardiometabolic health.25 Given that patients with AATD-associated severe COPD are generally younger than those with usual COPD,26 it is crucial to support this population to engage in physical activity.25
Pulmonary rehabilitation can help to improve QoL in patients with COPD.27 However, differences in muscle adaptation to pulmonary rehabilitation exercise regimens have been observed in patients with AATD-associated COPD versus those with usual COPD,28 and further research is needed to understand the impact of this intervention in patients with AATD.29
While high-intensity and moderate-intensity exercise training appear to be equally effective on exercise capacity, QoL, and dyspnoea in patients with AATD-associated COPD,28 high-intensity exercise training may be associated with greater benefits in terms of psychological comorbidities.30 This is an important consideration in this population, as anxiety and depression may be common in AATD.11,30,31
van ‘t Wout concluded that AATD is a heterogenous disease, and that treatment is partly dependent on phenotype and genotype. Clinicians should encourage physical activity in their patients with AATD-associated lung disease, as this can improve exercise capacity, QoL, and dyspnoea. van ‘t Wout also emphasised that treating physicians should not overlook the psychological impact of AATD.
Perspectives on Bronchiectasis
Bronchiectasis is defined as a chronic, abnormal dilation of the bronchi accompanied by classical symptoms.32,33 Typically caused by chronic airway inflammation and/or infection, the disease can be associated with multiple comorbidities, including airway diseases (asthma, COPD, chronic rhinosinusitis), cardiovascular disorders, and gastro-oesophageal reflux disease. Anxiety and depression are common comorbidities in bronchiectasis.31,32,34,35
The silent drivers of progression in bronchiectasis and the value of precision medicine in managing the disease were discussed by Michal Shteinberg, Clinical Associate Professor of Medicine, Technion, Israel Institute of Technology, and the Pulmonology Institute and CF Centre, Carmel Medical Centre, Haifa, Israel; and Sanjay H. Chotirmall, Associate Professor of Molecular Medicine, Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore.
Silent Drivers of Progression in Bronchiectasis: What are We Still Missing?
Shteinberg discussed how to classify the severity of bronchiectasis, how to decide on treatment goals, and how to determine the risk for complications.
What is severe bronchiectasis?
Disease severity is a reflection of the lung damage that bronchiectasis has already caused, measured mainly through a combination of FEV1, extent of radiological dilatation, and the presence of emphysema.36,37 Several different disease severity scores are used to assess bronchiectasis, including the Bronchiectasis Severity Index (BSI);38 FEV1, Age, Colonisation, Extension, Dyspnoea (FACED);39 and extended FACED (E-FACED).40 Disease severity scores classify bronchiectasis as mild, moderate, or severe, and can be used to predict mortality and hospitalisations.38-40 However, each system uses different parameters and different weighting.38-40 In addition, Shteinberg emphasised that severity scores are heavily influenced by age, and young people are therefore scored as having milder disease than older individuals with similar parameters.
Disease activity, on the other hand, is a reflection of current airway inflammation and can be used to predict disease progression.36,37 Common measures of disease activity include sputum purulence, frequency of exacerbations, severity of daily symptoms, neutrophil elastase concentrations, and blood or sputum eosinophil counts.36
The green colour in the sputum of patients with chronic inflammatory lung disease is thought to reflect the accumulation of myeloperoxidase, an enzyme released during neutrophilic inflammation.37 Sputum purulence can be used for the non-invasive assessment of disease activity in bronchiectasis, and to predict the future risk of exacerbations and mortality.41
The severity of daily symptoms can also be used to predict future pulmonary exacerbations.42 For example, real-world data from the EMBARC registry (N=9,466) showed that for each previous exacerbation, and for each 10-point reduction in QoL score, the rate ratios for future exacerbations were 1.11 (p<0.0001) and 1.10 (p<0.0001), respectively.42
What are our most impactful treatment goals?
Shteinberg explained that clinical trials in bronchiectasis most often demonstrate efficacy through achieving microbiology goals and reducing pulmonary exacerbations, but that improvements in lung function and QoL are less common, despite QoL being important to patients in real life.43,44 In addition, patients who do respond with FEV1 improvement often do not have a corresponding QoL improvement, and vice versa.45 Clinical trial data also demonstrate a significant placebo effect on QoL, with 26–56% of patients who receive placebo showing clinically important improvements in QoL.45
Who are the patients at high risk for complications?
In patients with bronchiectasis, comorbidities such as COPD, connective tissue disease, inflammatory bowel disease, and asthma are associated with a particularly high mortality risk.34 The Bronchiectasis Aetiology Comorbidity Index (BACI), in which comorbidities are weighted according to their impact on mortality, can be used to predict 5-year mortality rate, hospitalisation, exacerbations, and health-related QoL, and can inform decisions about which patients should be closely followed up.34 Comorbidity with rheumatoid arthritis occurs in 1–23% of patients with bronchiectasis, and is associated with more frequent exacerbations, higher disease severity, and elevated mortality compared to those without rheumatoid arthritis.46
Symptom duration and congenital aetiologies have been shown to correlate with disease severity in bronchiectasis, indicating that the longer a patient has been living with symptoms, the more severe the disease.47 Patients with paediatric-onset bronchiectasis in the EMBARC registry (n=249) were shown to have a longer disease duration, worse lung function, greater radiological extent, higher bacterial infection rate, and an increased exacerbation frequency compared to those with adult-onset bronchiectasis (n=1,173). Symptom duration was also shown to be independently associated with exacerbation frequency, hospitalisations, Pseudomonas infection, and lung function. Congenital aetiologies such as primary ciliary dyskinesia and primary immune deficiency also impacted disease severity.
Shteinberg emphasised the importance of looking for the silent drivers of severity alongside severity scores. This includes markers of disease activity such as sputum purulence, symptom score, and exacerbation frequency, and predictors of complications such as comorbidities, longer symptomatic disease duration, and congenital aetiologies.
Bronchiectasis: Is Precision Medicine the Answer?
There has been a significant increase in the number of global registries and clinical trials for bronchiectasis over the past 20 years,48 accompanied by an exponential growth in the quantity and quality of bronchiectasis research.49
Chotirmall explained that the key lesson learned is that bronchiectasis has a high degree of clinical heterogeneity, yet there remain several unanswered questions. For example, more research is needed to understand the fundamental origins, mechanisms, and natural history of bronchiectasis, potentially different types of the disease, whether any are reversible, and if some have origins in early life.
The understanding of the pathogenesis of bronchiectasis has evolved from a ‘vicious cycle’ of disease to a ‘vicious vortex’ model, in which infection, inflammation, and airway dysfunction intersect concurrently, rather than sequentially, with structural disease.35,50 The ‘vicious vortex’ model provides a framework to develop personalised pharmaceuticals for bronchiectasis, such as muco-active agents to treat airway dysfunction, anti-inflammatory agents to treat inflammation, and antimicrobial agents to treat infection.35 Chotirmall emphasised that in precision medicine, it is necessary to use the right intervention at the right time for each individual patient to get the best treatment response.
Multi-omics technologies, covering genomics, epigenomics, transcriptomics, metabolomics, and microbiomics, support the individualisation of therapeutic options through the analysis of integrated datasets, permitting the identification of the type and severity of disease, and predicting individual treatment response.51 Multi-omics endotype profiling (Figure 3) can offer the potential to identify novel therapeutic targets, as well as treatable and targetable traits in each individual patient with bronchiectasis.
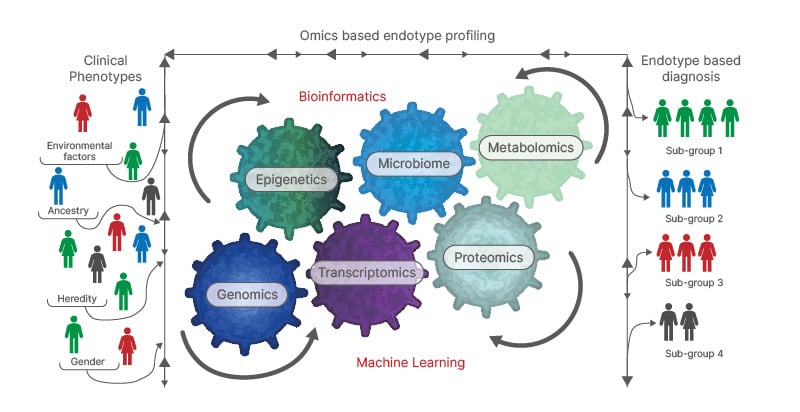
Figure 3: Identifying specific endotypes in bronchiectasis.
Adapted from Mersha et al.52
Chotirmall explained that once clinical disease traits have been identified at the individual patient level, and based on the ‘vicious vortex’ framework, disease targets can be prioritised. For example, a patient might have a clinical trait of frequent exacerbations, an inflammatory trait of neutrophilic inflammation, or a microbial trait of chronic Pseudomonas infection.35 Each of these characteristics can be treated with macrolides, dipeptidyl peptidase 1 inhibition, or eradication therapies, respectively; however, it is important to remember that once treatment is initiated, the same patient’s disease may evolve to develop different traits, and precision treatment will need to be reassessed.48
It is advisable to consider a broad range of clinical disease traits as having potential for treatment in clinical practice, since a complex intersection of disease traits exists in bronchiectasis, and targeting one aspect of the disease may have an unexpected impact on others. As an example, Chotirmall described a therapeutic agent currently in development, nebulised polyclonal human IgG, that demonstrated an exploratory endpoint of a reduction in bacterial sputum load in patients with non-cystic fibrosis bronchiectasis in a Phase I study (NCT04643587).53 A Phase II trial to further explore a potential therapeutic benefit with a primary endpoint of reduced exacerbations is ongoing (NCT07048262).54
Chotirmall summarised four steps in the personalised medicine approach to bronchiectasis:
- Understand that the disease presents with different forms and complexity in individual patients.
- Use unique algorithms to support a precision approach to different disease targets based on the ‘vicious vortex’ framework.
- Consider precision medicine an ongoing dynamic process that requires periodic reassessment.
- Continuously monitor the disease domains, severity (multi-dimensional scores), activity (sputum purulence, exacerbations, body weight), and impact (cough, dyspnoea, comorbidities), as part of a holistic precision medicine approach to management.
The Patient Voice: Living with Advanced Lung Disease
Thomas, a patient representative from Flensburg, Germany, discussed living with advanced lung disease from his own perspective. He was diagnosed with AATD at the age of 36 years, with an FEV1 of 70% at the time. Ten years later, his FEV1 had declined by 20%, prompting the initiation of ongoing augmentation therapy. Today, at the age of 65 years, Thomas’ FEV1 is 35%, constituting a 15% decline over nearly 20 years, which he feels shows that augmentation therapy has been beneficial.
Thomas explained that training is one of the most important ways to get the most out of his damaged lungs. He pursues three main training goals: strengthening his auxiliary muscles to maximise his lung capacity, increasing/maintaining his endurance to stabilise his overall physical condition, and general strength training to maintain muscle mass.
He also considers rehabilitation to be important. He, however, finds inpatient programmes to be more effective than outpatient programmes because they allow patients to focus intensively without having to cope with everyday life at the same time. Thomas has had four sessions of 3–4-week inpatient rehabilitation over the past 20 years, and uses the essential exercises and behaviours he has learned to help him through daily life. He also performs pulmonary rehabilitation techniques at his local gym and has regular connective tissue massages.
Thomas explained that he has a great deal of autonomy in managing his disease, including self-administration of his AAT therapy, and he is supported in this by his family doctor. He considers the optimal approach to be a combination of substantial patient autonomy and self-management, complemented by close collaboration with the family doctor in more challenging situations.
Key Takeaways
AATD is a heterogenous disease, and treatment partly depends on phenotype and genotype. While AATD is commonly associated with pulmonary manifestations such as emphysema,1,11 it is important to recognise that it can also present as liver disease6 or panniculitis.2 There is increasing evidence to support a substantial survival benefit of AAT augmentation therapy in patients with AATD,4,5 yet the association with FEV1 decline is unclear.4,22,23 To improve QoL in patients with AATD-associated lung disease, clinicians should encourage physical activity.25,30
The past 25 years have seen substantial growth in the understanding of bronchiectasis.48,49 We are entering an exciting period in the field, with the emergence of mature international registries, advances in multi-omics technologies and endotype-based classification, and new pipeline therapeutics (Chotirmall, personal communication). The coming years will likely see the emergence of a new era of precision medicine in bronchiectasis.