INTRODUCTION
Autoimmune polyendocrine syndrome Type 2 (APS-2) is a rare disorder characterised by progressive multi-glandular dysfunction, classically involving primary adrenal insufficiency, autoimmune thyroid disease, and Type 1 diabetes, and may occur alone or in association with other autoimmune disorders such as coeliac disease, alopecia, vitiligo, premature ovarian insufficiency (POI), and pernicious anaemia.1-3
Most causes of POI remain undefined; however, it is estimated that anywhere from 4–30% of cases are autoimmune in origin,4 having a strong relationship with other autoimmune conditions, primarily with autoimmune thyroid disease, followed by adrenal autoimmune disorders.5 In the context of APS-2, autoimmune POI is underestimated but clinically significant, with major consequences for fertility and long-term health.
CASE REPORT
The authors report the case of a 54-year-old woman with a complex form of APS-2. Autoimmunity first manifested at the age of 36 years as chronic autoimmune thyroiditis with hypothyroidism, with thyroid peroxidase antibodies 282 IU/mL and thyroid-stimulating hormone level 7 mIU/mL. Levothyroxine replacement therapy was initiated, achieving good clinical and biochemical control, as confirmed by thyroid stimulating hormone levels. At the age of 39 years, despite adequate treatment, the patient developed secondary amenorrhea, which was ultimately attributed to autoimmune POI, representing a key sentinel event in the course of her disease.
Several years later, at the age of 54 years, the patient presented with classical features of primary adrenocortical insufficiency, hypotension, fatigue, mucocutaneous hyperpigmentation, and increased craving for salt, raising clinical suspicion for Addison’s disease, which was confirmed by biochemical findings showing elevated adrenocorticotropic hormone levels and low serum cortisol concentration, hyponatraemia, and hyperkalaemia. Hormone replacement therapy was started with hydrocortisone and fludrocortisone, with improvement of clinical and biochemical results, serum sodium 141 mEq/mL and serum potassium 4.84 mEq/mL.
Subsequently, symptoms of hyperglycaemia developed in the context of adrenal hormone replacement therapy and weight gain, initially considered steroid diabetes, with C peptide value within the normal reference values. Antidiabetic therapy with metformin was initiated; however, glycaemic control progressively deteriorated. Based on the patient’s medical history and the coexistence of autoimmune disorders, it became necessary to exclude alternative causes of hyperglycaemia. Therefore, glutamic acid decarboxylase antibodies were assessed, and their positivity (glutamic acid decarboxylase 140 IU/mL) confirmed the diagnosis of Type 1 diabetes.
DISCUSSION
This case demonstrates the progressive and multifaceted nature of APS-2. The early appearance of autoimmune POI, predating other endocrine failures by over a decade, underlines the importance of recognising secondary amenorrhea in young women as a potential marker of autoimmune disease (Figure 1). Raising awareness among endocrinology and gynaecology teams regarding the diagnosis of POI and the assessment of an autoimmune substrate could be life-changing for the patient, potentially preventing an adrenal crisis, which may be fatal.
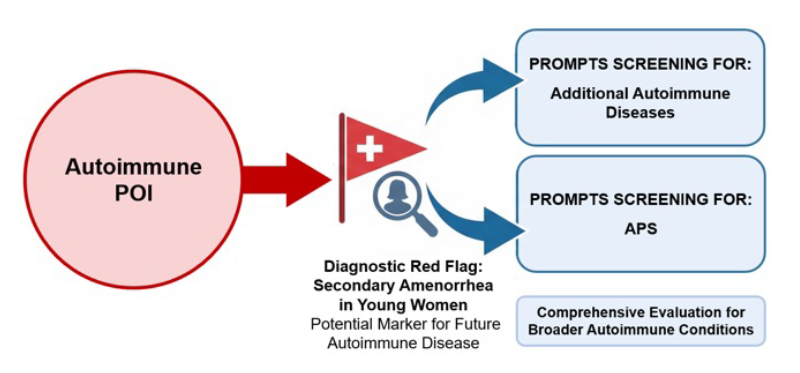
Figure 1: Autoimmune POI as a diagnostic red flag prompting screening for additional autoimmune diseases and APS.
APS: autoimmune polyglandular syndromes; POI: premature ovarian insufficiency.
CONCLUSION
APS-2 is a progressive, heterogeneous condition requiring long-term integrated management. Autoimmune POI may be the earliest clinical clue and should prompt active screening for associated endocrinopathies, especially measure of anti-21-hydroxylase antibodies and adrenal cortex autoantibodies, to rule out Addison’s disease.6 Early recognition and personalised treatment strategies, encompassing hormone replacement, fertility counselling, and careful metabolic management, are critical for improving outcomes.