Abstract
Sjögren’s Disease (SjD) is a chronic autoimmune disorder that predominantly affects women but may also occur in men and children, often with distinct clinical presentations. This review aimed to describe the main clinical findings, complications, and therapeutic approaches of SjD in these two underrepresented populations and to compare them with the classical phenotype observed in adult women. A total of 117 studies were analysed, comprising 74 adult male cases and 65 paediatric patients. In men, glandular manifestations were frequent but often incomplete, with isolated oral and/or ocular symptoms associated with significant systemic involvement, particularly neurological (38.2%), pulmonary (31.1%), and nephro–urological (21.6%) complications. In children, enlargement of the major salivary glands was the most common manifestation (75.4%), followed by musculoskeletal (44.6%), haematological (44.6%), cutaneous (29.2%), renal (20.0%), and neurological (13.8%) involvement. Complete sicca syndrome was less frequent in both groups; however, isolated glandular symptoms and early systemic manifestations were common and clinically relevant. SjD in men and paediatric patients demonstrates greater clinical heterogeneity and a higher burden of systemic manifestations compared with the classical adult female phenotype, underscoring the need for heightened clinical awareness and tailored diagnostic strategies in these populations.
Key Points
1. Although Sjögren’s disease predominantly affects middle-aged women, adult men and juvenile patients represent underrecognised populations with distinct clinical characteristics.2. This review analysed 117 published studies to characterise the clinical presentation, systemic involvement, and diagnostic challenges of Sjögren’s disease in adult men and juvenile patients.
3. Adult men and juvenile patients frequently present with incomplete sicca syndrome and early systemic manifestations, highlighting the need for tailored diagnostic strategies and earlier recognition.
INTRODUCTION
Sjögren’s disease (SjD) derives its name from the Swedish ophthalmologist Henrik Samuel Conrad Sjögren, who, in 1933, was the first to describe the association between keratoconjunctivitis sicca, xerostomia, and polyarthritis.1 SjD is a chronic, systemic autoimmune disorder of unknown aetiology characterised by dysfunction of the salivary and lacrimal glands, resulting in oral dryness (xerostomia) and ocular dryness (xerophthalmia). Dryness may also affect other mucosal surfaces, including the respiratory tract, gastrointestinal tract, and vagina, resulting in a broader spectrum of sicca manifestations beyond the salivary and lacrimal glands.2
In addition to glandular involvement, SjD presents with systemic manifestations affecting multiple organs, such as arthritis, interstitial pneumonitis, interstitial nephritis, isosthenuria or renal tubular acidosis, thyroiditis, involvement of the central, peripheral, and autonomic nervous systems, vasculitis, and an increased risk of lymphoma.3 Throughout this review, the term ‘extraglandular manifestations’ refers specifically to systemic organ involvement outside the exocrine glands.
SjD may occur as an isolated condition or in association with other autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus.4,5 Although SjD may occur at any age, approximately two-thirds of cases are diagnosed between 30–60 years of age, predominantly affecting middle-aged women, with an estimated prevalence ranging from 0.01–0.72%. In children, SjD is less well characterised in terms of clinical presentation and long-term outcomes.5,6
The disease is rare in childhood and may be underrecognised. When onset occurs before the age of 18 years, it is referred to as early-onset, childhood, or juvenile SjD.7 Clinical manifestations in children may differ from those observed in adults, such as recurrent parotid enlargement. However, pathological and laboratory findings are similar to those in adults, including characteristic lymphocytic infiltration of exocrine glands, hypergammaglobulinemia, elevated erythrocyte sedimentation rate, positivity for anti-Ro and anti-La antibodies, antinuclear antibodies (ANA), and rheumatoid factor.8,9
With regard to male patients, few cases have been reported in the literature describing their clinical characteristics, complications, and therapeutic approaches. The female-to-male ratio may reach up to 20:1 in Asian populations, although this proportion varies across different regions.10,11
Therefore, this study aimed to conduct a review of the English language literature addressing the clinical characteristics and associated complications of SjD in male patients and children, in order to identify key differences and unique features compared with the typical manifestations observed in adult women.
METHODOLOGY
The present study consists of a narrative literature review with a qualitative and descriptive approach, conducted with the aim of gathering, analysing, and synthesising the available scientific evidence regarding SjD in male patients and in the paediatric population. This design was chosen to allow for a broad, critical, and interpretative analysis of published findings, enabling clinical, diagnostic, and therapeutic contextualisation of the cases described in the literature.
The bibliographic search was conducted through electronic databases like PubMed, ScienceDirect, Web of Science, Embase, and Scopus, without restriction on the initial publication date, including studies published up to January 2026. Additionally, a complementary search was performed using Google Scholar, considering the first 100 results ranked by relevance. Controlled descriptors and free-text terms were combined using Boolean operators according to the following search strategy: (“Sjögren’s syndrome” OR “Sjögren Syndrome” OR “Sicca Syndrome”) AND ([male OR man] OR [child OR children]) AND (“Complications” OR “associated disease” OR “Coexistent Disease” OR “Associated conditions” OR “Concomitant disease”). The strategy was adapted according to the indexing characteristics of each database to enhance search sensitivity and comprehensiveness.
Case reports, case series, and observational studies published in English that described male and/or paediatric patients with SjD and that provided sufficient clinical, laboratory, and/or histopathological information to support the clinical diagnosis were included. Patients were considered eligible when they fulfilled the 2016 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for SjD, which were used for research classification purposes rather than as diagnostic criteria. As clinical diagnosis of SjD relies on expert clinical assessment and should be distinguished from research classification, studies were included if patients met these criteria and the diagnosis was adequately described by the authors. For studies originating from Asian countries, diagnoses based on the 1999 Japanese criteria, which are considered diagnostic criteria, were also accepted, provided they were clearly described. Studies with inadequate or inconclusive diagnostic criteria; publications lacking sufficient information to support the clinical diagnosis; exclusively laboratory-based, immunohistochemical, histomorphometric, gene expression, or in vitro experimental studies without detailed clinical descriptions; review articles; and publications in languages other than English were excluded.
Study selection was initially performed through screening of titles and abstracts identified in the databases, followed by full-text assessment of potentially eligible articles. In cases of uncertainty regarding diagnostic adequacy or study eligibility, a careful analysis of the methodological and clinical information provided was conducted, considering the consistency of the reported data and adherence to recognised diagnostic criteria.
Data from the included studies were extracted in a standardised manner and organised in an electronic spreadsheet developed using Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA). The variables collected included author and year of publication, country of origin, total number of patients, sex, age at diagnosis, disease duration, initial clinical manifestations, glandular and extraglandular signs and symptoms, associated complications, diagnostic criteria used, laboratory and serological findings, results of minor salivary gland biopsy, including the presence of lymphocytic foci when available, therapeutic approaches employed, follow-up duration, and clinical outcome.
The collected data were analysed descriptively, with emphasis on clinical characterisation and associated complications. The synthesis was conducted in a narrative and interpretative manner, aiming to identify recurrent clinical patterns, diagnostic particularities, and potential differences in disease presentation between male and paediatric patients, thereby contributing to a more comprehensive understanding of these less frequently described manifestations of SjD.
RESULTS
The results are summarised in (Table 1).
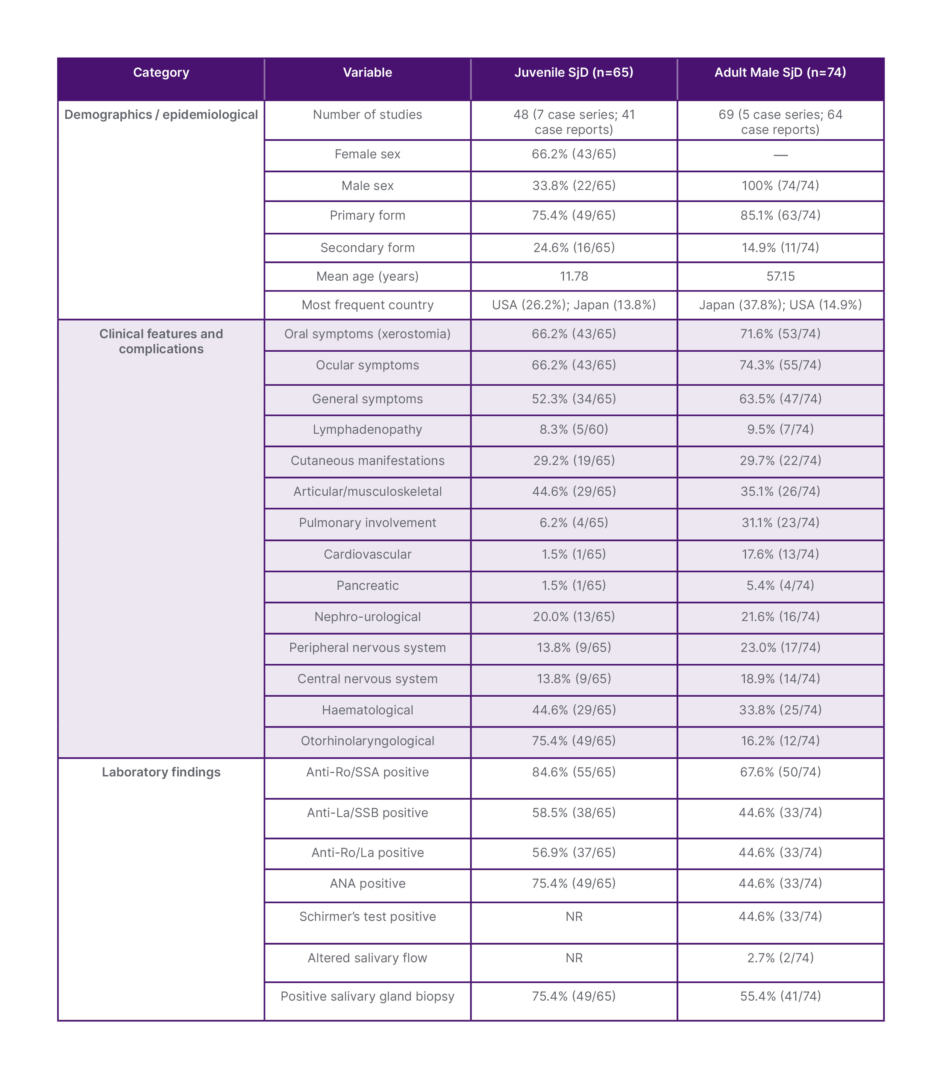
Table 1: Demographic, clinical, and laboratory characteristics of juvenile and adult male SjD.
ANA: antinuclear antibodies; Anti-La: anti-La antibodies; Anti-Ro: anti-Ro antibodies; NR: not reported;
SjD: Sjögren’s disease.
The literature search identified 2,943 records from electronic databases and indexed sources, and 100 from gray literature. After the removal of 91 duplicates, 2,952 records underwent title and abstract screening. Of these, 2,416 were excluded for irrelevance and 59 (including seven from Google Scholar) were excluded due to unavailable full texts, resulting in 477 articles assessed for eligibility. Following full-text evaluation, 408 were excluded due to inappropriate study design (n=78), publication type (n=58), non-English language (n=37), inadequate population (n=203), or insufficient diagnostic/clinical information (n=32). Consequently, 69 studies were included in the adult male analysis (50 from databases and 19 from gray literature).
For the paediatric population, 2,916 records were identified. After removing 94 duplicates, 2,822 records were screened, yielding 367 potentially relevant articles. Full-text review led to exclusion of 319 studies due to inappropriate design (n=46), publication type (n=77), foreign language (n=32), inadequate population (n=82), insufficient information (n=45), or failure to meet inclusion criteria (n=37). A total of 48 studies were included in the paediatric subgroup analysis.
Study Characteristics Adult male population
Sixty-nine studies (64 case reports and five case series) comprising 74 patients were analysed. Isolated SjD was identified in 63 cases, whereas SjD associated with another autoimmune disease was identified in 11 cases. The mean age was 57.15 years. Japan contributed 30 studies and the USA 12, with an additional 14 countries reporting isolated cases.
Juvenile population
Forty-eight studies (41 case reports and seven case series) included 65 patients. Females represented 66.2% and males 33.8% (approximately a 2:1 ratio). Isolated SjD was present in 49 cases, whereas SjD associated with another autoimmune disease was identified in 16 cases. The USA contributed 27 publications and Japan 14, with the remaining cases distributed across Europe, Asia, and Latin America. The mean age was 11.78 years.
Clinical Features and Complications Adult male population
Oral manifestations occurred in 53 patients, predominantly xerostomia, frequently associated with mucosal alterations and salivary gland enlargement. Ocular involvement was observed in 55 patients, mainly xerophthalmia. Complete sicca syndrome was reported in 42 patients, whereas 11 presented isolated oral symptoms and 13 isolated ocular symptoms.
General symptoms were reported in 47 patients, most commonly fever (approximately 18 cases) and fatigue/asthenia (approximately 16 cases). Respiratory symptoms (cough and dyspnoea) occurred in approximately 15 cases. Lymphadenopathy was identified in 9.5%.
Cutaneous manifestations were present in 22 patients, mainly xerosis and erythematous eruptions; purpura, vasculitis, oedema, and dermatomyositis were less frequent. Musculoskeletal involvement occurred in 26 cases, predominantly arthralgia and joint pain, followed by muscle weakness and myalgia; severe cases included myopathy and atrophy.
Cardiovascular complications were observed in 13 patients, mainly hypertension and orthostatic hypotension; less frequent findings included Raynaud’s phenomenon, vasculitis, inflammatory periaortitis, constrictive pericarditis, and one case of acute eosinophilic myocarditis with cardiogenic shock.
Nephro-urological involvement occurred in 16 patients (21.6%), mainly glomerular disease (proteinuria ± hematuria), pauci-immune glomerulonephritis, and severe proteinuria. Tubulointerstitial disease included chronic tubulointerstitial nephritis, renal tubular dysfunction, distal renal tubular acidosis, nephrocalcinosis, hypokalaemia, and hypokalaemic paralysis. One patient required renal transplantation. Lower urinary tract symptoms were less frequent.
Neurological involvement was common: peripheral nervous system involvement occurred in 23% (mainly sensory neuropathy), and central nervous system involvement in approximately 19% (ataxia, encephalopathy, autoimmune encephalitis, limbic encephalitis, cerebral vasculitis, demyelinating disease, hemiplegia, language disturbances, and cerebrovascular events).
Pancreatic complications were reported in four patients. Haematological abnormalities occurred in 25 of 74 patients (33.8%), including anaemia, leukopenia, thrombocytopenia, pancytopenia, hypergammaglobulinemia, elevated IgG, cryoglobulinemia, hypocomplementemia, and elevated inflammatory markers. T cell lymphoma and MALT lymphoma were rarely reported. Otorhinolaryngological manifestations were observed in 12 patients (16.2%), mainly parotid enlargement.
Juvenile population
Oral symptoms were present in 43 patients (xerostomia in 37), and ocular symptoms in 43 (xerophthalmia in 32). General symptoms were reported in 52.3% of cases, and lymphadenopathy in approximately 8%.
Otorhinolaryngological involvement was the most frequent extraglandular manifestation (75.4%), particularly parotid enlargement (approximately 46 patients), often bilateral and recurrent. Submandibular enlargement occurred in about seven cases, and recurrent/chronic parotitis in approximately eight; structural abnormalities were less frequent. Thyroid involvement was rare.
Musculoskeletal manifestations occurred in 29 patients, mainly arthralgia and polyarticular inflammatory arthritis; muscle weakness and osteopenia were less common. Haematological abnormalities were also reported in 29 patients, primarily elevated erythrocyte sedimentation rate/CRP (approximately 18 cases) and hypergammaglobulinemia (approximately 16 cases). Anaemia, cytopenias, hypocomplementemia, and haematologic malignancies were rare.
Cutaneous involvement occurred in 19 patients. Renal involvement was reported in 13 cases (20%), mainly proteinuria and membranous glomerulonephritis. Neurological involvement occurred in nine cases, pulmonary in four, and cardiovascular and pancreatic complications in one case each.
Laboratory Findings
Among adult men, anti-Ro antibodies were positive in 50 patients (64.1%), anti-La antibodies in 33 (42.3%), and combined anti-Ro/anti-La positivity was observed in 44.6% of cases. ANA positivity was identified in 45.5% of patients, and Schirmer’s test was positive in a similar proportion. Rheumatoid factor was positive in 22% of cases when reported. Minor salivary gland biopsy showed positive findings in 55.4% of patients.
In juvenile patients, anti-Ro positivity reached 84.6% (55/65), anti-La antibodies were identified in 38 cases, and combined anti-Ro/anti-La positivity occurred in 37 cases. ANA was positive in 75.4% (49 patients), and minor salivary gland biopsy was positive in 49 patients.
Risk of Bias
Among adult male studies, 45 of 64 case reports were low risk, 15 moderate, and four high; of five case series, two were low risk and three moderate. In the juvenile group, 39 of 41 case reports were low risk and two moderate; among seven case series, five were low risk, one moderate, and one high.
DISCUSSION
SjD is a chronic, systemic autoimmune connective tissue disorder mediated by B and T lymphocytes, primarily affecting the salivary and lacrimal glands. Clinically, it is characterised by sicca manifestations, with xerostomia and xerophthalmia as the central features.1 The disease demonstrates a marked female predominance, with a reported female-to-male ratio of approximately 12:1 in different population-based studies.12,13 This epidemiological profile has contributed to the fact that most of the available knowledge regarding SjD derives from cohorts composed predominantly of adult women, in whom glandular manifestations are present in more than 90% of cases and complete sicca syndrome is identified in approximately 89% of patients. This may limit the understanding of the disease in other populations.14
In this context, the present review aimed to synthesise and compare the demographic, clinical, and laboratory characteristics of SjD in two historically underrepresented populations: juvenile patients and adult men. Although previous reviews have addressed these groups separately, the comparative approach proposed in this study allows the identification of distinctive clinical patterns and diagnostic challenges that are not fully characterised when relying exclusively on the classical model based on adult female cohorts, particularly regarding the frequency of incomplete sicca presentations and the predominance of systemic manifestations.
This review adopted the two most widely used sets of criteria for SjD: the 2016 ACR/EULAR classification criteria and the Japanese diagnostic criteria (1999). The main differences between them lie in the inclusion of anti-La antibodies and lacrimal gland biopsy in the Japanese diagnostic criteria, whereas these items are not included in the 2016 ACR/EULAR classification criteria. In the latter, positivity for anti-Ro antibodies or a minor salivary gland biopsy with a focus score >1 is required for classification.15,16 It should be emphasised that the 2016 ACR/EULAR criteria were developed for research classification purposes and are not intended to establish a clinical diagnosis of SjD.
Based on the classification criteria applied in this study, 74 adult male patients who fulfilled the inclusion criteria were analysed, predominantly derived from case reports and small case series, with a mean age of 57.15 years and a clear predominance of isolated SjD. A broad geographic distribution was observed, with the highest concentration of cases reported in Japan and the USA, reflecting both disease prevalence and potential publication bias due to underreporting. Clinically, glandular manifestations were frequent, with ocular symptoms in 55 patients and oral manifestations in 53, and complete sicca syndrome identified in 42 individuals. Nevertheless, a proportion of patients presented isolated glandular symptoms, with 11 reporting only xerostomia and 13 only xerophthalmia, indicating that incomplete presentations also occur in men and may contribute to diagnostic delay in selected cases.
The occurrence of SjD in children and adolescents is considered rare, accounting for approximately 1% of patients.7 In this population, 65 patients were analysed, with a mean age of 11.78 years and a female predominance of approximately 2:1. As observed in adult men, most patients had isolated SjD rather than SjD associated with another autoimmune disease. The USA and Japan accounted for the majority of publications, with additional cases reported in other countries across Europe, Asia, and Latin America.
In young patients, initial signs and symptoms are often nonspecific and may mimic other autoimmune or systemic inflammatory disorders, rendering diagnosis challenging and frequently delayed.17 The authors’ findings reflect this complexity, as despite the high frequency of glandular manifestations, complete sicca syndrome occurred in a lower proportion of cases, while isolated xerostomia or xerophthalmia was observed in approximately 23.3% of patients. Moreover, enlargement of major salivary glands, particularly parotid and submandibular glands, was identified in up to 91.6% of juvenile patients, representing the most frequent clinical sign in this group. A substantial burden of extraglandular manifestations was also observed, including musculoskeletal, neurological, renal, and cutaneous involvement, suggesting that in the juvenile population SjD may present as a systemic disease from early stages, frequently preceding or masking classical sicca symptoms.
When analysed comparatively, the data from this review suggest that SjD exhibits greater clinical heterogeneity in juvenile patients and adult men than that traditionally described in adult female cohorts. While in the latter, glandular manifestations and complete sicca syndrome are considered predominant clinical features, in juvenile and male populations, a greater variability in presentation was observed, with a relevant proportion of cases characterised by incomplete sicca symptoms and early systemic involvement.2,12,14 This pattern may contribute to additional diagnostic challenges in these groups, particularly when initial clinical suspicion of SjD is low.
In the juvenile population, extraglandular manifestations, particularly musculoskeletal and neurological, were frequent. Musculoskeletal complaints included arthralgia in approximately 15% of patients and arthritis in 6.6%, while cutaneous manifestations were observed in about 30% of paediatric cases. This profile reinforces the need to consider SjD in the differential diagnosis of children and adolescents presenting with recurrent systemic manifestations associated with autoimmune findings, even in the absence of overt sicca complaints.
Conversely, adult men demonstrated a higher frequency of pulmonary, renal, and peripheral neurological involvement. Pulmonary involvement was identified in approximately 29.4% of cases, while neurological manifestations occurred in 38.2%, predominantly in the form of painful sensory neuropathy and cranial neuropathies. Renal involvement was also significant, present in approximately 20.5% of patients, with proteinuria and tubular dysfunction as the predominant manifestations. These findings may reflect delayed diagnosis in this group, in whom SjD is often recognised only after systemic manifestations become apparent. Additionally, an increased prevalence of Klinefelter syndrome has been reported among men with SjD, supporting the hypothesis that sex chromosome abnormalities may contribute to disease susceptibility. Some authors have even suggested that men with SjD or systemic lupus erythematosus who have not fathered children should be considered for screening for Klinefelter syndrome. Furthermore, men with SjD remain underrepresented in clinical studies, although evidence suggests a higher prevalence of neuropathy, fatigue, interstitial lung disease, and rheumatologic comorbidities, with greater functional impact compared to women.18,19
From a laboratory perspective, although anti-Ro positivity was high in both groups, its diagnostic relevance appears particularly significant in the juvenile population, in whom classical clinical criteria are often not fully met. It should be noted that, although current nomenclature recommends distinguishing between anti-Ro52 and anti-Ro60 antibodies, the data available in the included studies only allowed the identification of anti-Ro positivity. In juvenile SjD, anti-Ro antibodies were detected in approximately 81.6% of cases and ANA in about 80%, even in the absence of complete sicca manifestations. In contrast, in adult men, there was greater heterogeneity in the performance and reporting of complementary tests, with anti-Ro positivity in approximately 64.7%, ANA positivity in 45.5%, and rheumatoid factor in 22% of cases, suggesting less diagnostic standardisation compared to female cohorts.
Among the main strengths of this review are the direct comparative approach between two underrepresented populations, the systematic integration of demographic, clinical, and laboratory data, and the updating of the current body of evidence. However, inherent limitations must be acknowledged. The analysed data in both juvenile patients and adult men were derived exclusively from case reports and small case series, limiting generalisability and increasing susceptibility to publication bias, with possible overrepresentation of severe or atypical presentations. Additionally, incomplete and heterogeneous reporting of clinical and laboratory data restricted the possibility of more robust quantitative analyses. Therefore, the findings should be interpreted with caution, reinforcing the need for prospective, multicentre studies applying standardised diagnostic criteria to better characterise SjD in these populations.
In summary, although SjD is classically associated with adult women, it presents relevant clinical and laboratory manifestations in children, adolescents, and men, often with distinct profiles, early systemic involvement, and greater diagnostic complexity. Recognition of these differences is essential to reduce diagnostic delays and optimise clinical management. Future studies, preferably multicentre and employing standardised diagnostic criteria, are necessary to further elucidate the natural history of SjD in these groups and to guide more precise therapeutic strategies.
CONCLUSION
The findings of this review contribute to the characterisation of SjD in less-studied populations, such as adult men and juvenile patients, by highlighting specific clinical and laboratory profiles that influence diagnostic recognition and disease management in these groups.
In adult men, glandular manifestations were frequent, although complete sicca syndrome was not consistently present. A high frequency of systemic manifestations was observed, including pulmonary, cutaneous, musculoskeletal, neurological, and nephro-urological involvement, as well as haematological complications. From a laboratory perspective, high positivity rates for anti-Ro and anti-La antibodies were identified, reinforcing their diagnostic relevance in this population.
In the juvenile population, SjD exhibited a distinct profile, with frequent glandular manifestations but less consistent presentation of complete sicca syndrome. Otorhinolaryngological alterations, particularly those associated with salivary gland enlargement, were especially prevalent. Extraglandular manifestations, including musculoskeletal, cutaneous, neurological, and renal involvement, were also common, as were haematological abnormalities. Serological evaluation demonstrated high positivity rates for ANA and anti-Ro antibodies.
Taken together, these results indicate that SjD in adult men and juvenile patients presents greater clinical heterogeneity and a higher burden of systemic manifestations compared with the classical phenotype described in adult women, underscoring the need for tailored diagnostic strategies in these populations.