Meeting Summary
At a non-promotional AstraZeneca-sponsored symposium at the European Respiratory Society (ERS) 2025 Congress, five experts were invited to discuss the current unmet need for biologic therapy in COPD, explore the roles of IL-33 in COPD pathogenesis, and review newly developed biologics for COPD that target IL-33 pathways. Andrew Menzies-Gow, Vice President, Respiratory & Immunology, Global Biopharmaceuticals Medical, AstraZeneca, Cambridge, UK, highlighted barriers to effective COPD management and emphasised the need for proactive, integrated, patient-centred care. Rebecca D’Cruz, Pulmonologist, Guy’s and St Thomas’ NHS Foundation Trust, London, UK, explained why novel COPD therapies are required and addressed the role of mucus dysfunction in COPD progression. Dave Singh, Pulmonologist and Clinical Pharmacologist, University of Manchester and Medicines Evaluation Unit, UK, described how the reduced (IL-33RED) and oxidised (IL-33OX) forms of IL-33 act through distinct pathways to promote inflammation, mucus dysfunction, and impaired epithelial repair in COPD. Stephanie Christenson, Pulmonologist, University of California, San Francisco, USA, summarised aspects of clinical trials evaluating IL-33-targeted biologics for COPD. Finally, Claus Vogelmeier, Pulmonologist, University Hospital Marburg, Germany, chaired a panel discussion of how targeting distinct IL-33 pathways might change the future landscape of COPD.
Introduction
Adequate management of COPD is important because hospitalisation for an acute exacerbation of COPD (AECOPD) is associated with high rates of pooled 365-day hospital readmission (38.2%), in-hospital mortality (6.2%), and pooled 365-day post-discharge mortality (12.2%).1 However, despite optimisation of maintenance treatment with combination inhalers, many patients continue to experience exacerbations,2 highlighting an unmet need for additional treatments. Biologics targeting immune system components, including IL-33 signalling, have been developed as potential new COPD therapies.3
The main objectives of the symposium described in this article were to raise awareness of the current unmet need for biologic therapy in COPD despite optimised standard-of-care treatment, explore the roles of IL-33RED and IL-33OX in COPD pathogenesis, and review new biologics for COPD that target IL-33 pathways.
Transforming Care in COPD
Andrew Menzies-Gow
Globally, around 468 million people lived with chronic respiratory diseases in 2021.4 Lung disease is a major driver of health inequalities.5,6 The number of deaths, disability-adjusted life years, and hospitalisations due to chronic respiratory diseases has increased during the past 3 decades.4,7,8 Healthcare systems have similar barriers to care for chronic respiratory diseases, including delayed diagnosis and fragmented care pathways.9-11 As a result, many individuals with chronic lung diseases are undiagnosed or undertreated.12,13 The main stakeholders in healthcare delivery, including healthcare providers, pharmaceutical companies, professional societies, and patient advocacy groups, need to work together to provide proactive, integrated, patient-centred care for chronic respiratory diseases, as early diagnosis and initiation of guideline-directed medical therapy decreases the rates of AECOPD and hospital admissions, and may reduce the rate of premature deaths.
The Unmet Need for Novel Therapies in COPD
Rebecca D’Cruz
COPD affects millions of people worldwide, but many patients receive suboptimal therapy and continue to experience moderate-to-severe exacerbations.14 Exacerbations have profound effects on patients’ disease progression,15 their physical and mental wellbeing,16,17 and their caregivers’ lives.18
A retrospective USA cohort study (SIRIUS I) included 4,920 patients with COPD on triple therapy with a history of ≥2 moderate or ≥1 severe exacerbations per year.14 During the first year of follow-up, 69% of participants experienced ≥1 moderate and/or severe exacerbation, while 25% of patients experienced ≥1 severe exacerbation requiring hospitalisation for ≥2 days.19 These arresting statistics highlight COPD as a devastating disease with serious implications.
Approximately 90% of patients in the SIRIUS I study received an oral corticosteroid (OCS) during baseline for a mean cumulative duration of 73 days.14 OCS exposure is associated with significantly elevated risks of various adverse outcomes, including pneumonia (adjusted hazard ratio [aHR]: 2.90; 95% CI: 2.77–3.03 versus no exposure), osteoporosis (HR: 1.80; 95% CI: 1.70–1.92), Type 2 diabetes (HR: 1.44; 95% CI: 1.37–1.51), and cardiovascular/cerebrovascular disease (HR: 1.26; 95% CI: 1.21–1.30).20 Notably, relative all-cause mortality rates were 74% higher for patients exposed to cumulative OCS doses of 0.5–<1.0 g (aHR: 1.74; 95% CI: 1.65–1.83) and 145% higher for patients exposed to cumulative OCS doses of 1.0–<2.5 g (aHR: 2.45; 95% CI: 2.33–2.58), in comparison to those exposed to <0.5 g of OCS.20
D’Cruz emphasised that patients with COPD frequently die of cardiovascular disease, and the management of cardiopulmonary risk in patients with COPD remains suboptimal. An AECOPD increases the risks of subsequent exacerbations and cardiovascular events, both of which are associated with premature death.21,22 A retrospective UK cohort study of 213,466 patients with COPD concluded that there was an approximately two-fold increase in the risk of acute coronary syndrome (aHR: 2.07; 95% CI: 1.39–3.09) and nearly three-fold elevations in the risks of arrhythmia (aHR: 2.86; 95% CI: 2.36–3.47) and heart failure (aHR: 2.87; 95% CI: 2.36–3.50) during the first 14 days after a moderate/severe exacerbation.23 Even a single moderate exacerbation may increase the risk of future exacerbations and is associated with a higher risk of premature mortality. In an observational analysis of 340,515 patients with COPD in the UK, one moderate exacerbation was associated with a 17% increase in the adjusted incidence rate ratio (aIRR) for COPD-related death (1.17; 95% CI: 1.04–1.33 versus no exacerbation) and a 23% higher aIRR for cardiovascular-related death (1.23; 95% CI: 1.07–1.42).24 Notably, the risks were even greater after one severe exacerbation, with aIRR increases of 138% for COPD-related death (2.38; 95% CI: 2.08–2.73 versus no exacerbation) and 65% for cardiovascular-related death (1.65; 95% CI: 1.34–2.02).24
Patients with COPD continuing to experience exacerbations on triple therapy have a severe and under-recognised disease burden. In an analysis of data (drawn from an international cross-sectional study) for 399 patients on triple therapy with productive cough and ≥2 moderate/≥1 severe exacerbations in the prior year, 54% exhibited severe-to-very-severe airway obstruction, 78% had breathlessness with a Modified Medical Research Council (mMRC) Dyspnoea Scale score ≥2, and 35% required O2 therapy.25,26 Unfortunately, the devastating implications of COPD are under-recognised by clinicians: a recent survey revealed that 73% of physicians considered their patients’ COPD to be somewhat/well/completely controlled, despite these patients exhibiting exacerbations on triple therapy.26 This highlights an incongruence between patients’ experiences and clinicians’ perceptions.
The Role of Mucus Dysfunction in COPD Progression
Rebecca D’Cruz
Mucus dysfunction is central to COPD pathology and includes mucus hypersecretion and mucus plugging.27-31 Mucus hypersecretion is associated with dysregulation of basal cell differentiation, mucin-5AC (MUC5AC) overproduction by bronchial epithelial goblet cells, impaired ciliary clearance, airway infection, and symptoms such as productive cough and dyspnoea.27,28 Viscous mucus plugs obstruct airways to increase airway resistance and drive hyperinflation, which raises the work of breathing and manifests as breathlessness.30,32,33 Mucus plugs in CT scans are associated with accelerated lung function decline,34 an elevated risk of exacerbations,34 and increased all-cause mortality.29
The COPDGene study is an observational prospective cohort study that included analysis of 4,363 patients with COPD at 21 centres in the USA.35 This analysis revealed that 74.5% of participants had signs of mucus dysfunction (cough, phlegm, and/or CT-detected mucus plugs).35 Interestingly, only approximately 35% of patients with mucus dysfunction had cough and/or phlegm as well as mucus plugs. Approximately 45% of participants had cough and/or phlegm only (indicative of mucus hypersecretion), while approximately 20% of participants had silent mucus plugs without cough or phlegm (Figure 1).35 This suggests that although mucus dysfunction is common in patients with COPD, mucus hypersecretion and mucus plugs may be distinct, and there is a poor correlation between the two.
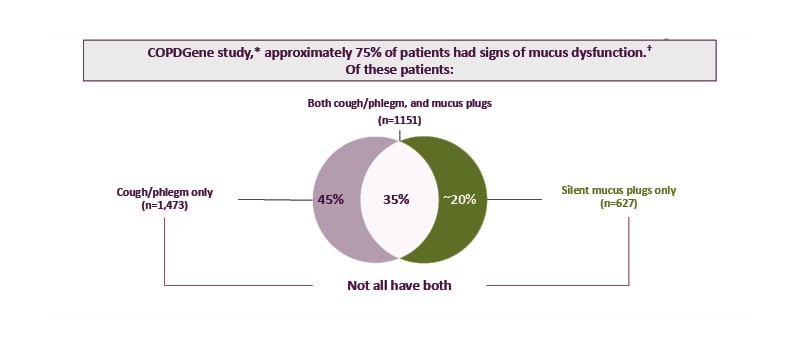
Figure 1: Mucus dysfunction is common in COPD, with a poor correlation between mucus hypersecretion and mucus plugs.
*Based on data from 4,363 patients with COPD (current or former smokers) across the full spectrum of COPD severity. Patients were recruited from the COPDGene study, an observational prospective cohort study conducted across 21 centres in the USA, and included 45–80-year-old non-Hispanic White or non-Hispanic Black patients with COPD and with a ≥10 pack-year smoking history.29,35
†Signs of mucus dysfunction included cough, phlegm, and/or mucus plugs.
Adapted from Mettler et al.,35 licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Productive cough and excess sputum are associated with numerous negative outcomes, including airflow limitation, exacerbations, dyspnoea, fatigue, physical activity limitation, depression, anxiety, and social isolation.36-39 Mucus plugs occlude the lumen of the airways.31,32,40 Furthermore, there is evidence that mucus plugs are more common in severe COPD: the prevalence of ≥1 mucus plug in CT images of 18 lung segments increased progressively from 22.1% in patients with mild COPD (Global Initiative for Chronic Pulmonary Obstructive Disease [GOLD] Stage I) to 63.1% in patients with very severe disease (GOLD Stage IV).29,41
Although some patients with COPD exhibit resolution of mucus plugging, others have persistent plugs or develop new mucus plugs. In a subgroup of 2,118 patients with COPD in the COPDGene study, who were followed up for 5 years including CT and spirometry assessments, the annual mean decline in forced expiratory volume in 1 second was faster in participants with persistent (60.4 mL/year) or newly formed (54.9 mL/year) mucus plugs than in those with resolved mucus plugs (39.3 mL/year) or absent mucus plugs (reference group; 37.2 mL/year).42 There is evidence that mucus plugs are associated with elevated risks of AECOPD and death. For example, a retrospective observational study of 374 propensity-score-matched patients with COPD demonstrated that CT-detected mucus plugs significantly increased the risk of moderate-to-severe exacerbations by 50% (aHR: 1.50; 95% CI: 1.12–2.02) and the risk of severe exacerbations by 111% (aHR: 2.11; 95% CI: 1.43–3.10 ) versus no mucus plugs.34 Furthermore, an observational retrospective analysis of COPDGene data found that the presence of mucus plugs was associated with significantly higher hazards of all-cause mortality: aHR 1.15 (95% CI: 1.02–1.29) for plugs in 1–2 lung segments and aHR 1.24 (95% CI: 1.10–1.41) for plugs in three or more segments versus none.29
Many patients with COPD continue to experience exacerbations on triple therapy, indicating an unmet need for novel therapies that target the broader mechanisms underlying COPD pathogenesis.43-46
The Distinct Pathways of IL-33 Driving COPD Pathogenesis
Dave Singh
IL-33-mediated signalling is a key pathway driving COPD pathogenesis. IL-33 is highly expressed in lung tissue homogenate from patients with severe COPD: IL-33 levels were significantly higher in patients with GOLD Stage III/IV COPD (forced expiratory volume in 1 second: <50% predicted) than in healthy controls (p<0.001).41,47 IL-33 levels correlate with an increased risk of future COPD exacerbations and prevalence of productive cough.48,49 In a prospective study of 62 patients with COPD in Korea followed for 1 year, participants in the highest quartile of plasma IL-33 concentration had a significantly higher exacerbation rate than patients with lower IL-33 levels (1.00±1.16 versus 0.40±0.62 exacerbations/year; p=0.01).48 Furthermore, a multicentre study of 307 people with COPD observed that productive cough (phlegm for ≥3 months/year) was significantly more prevalent in those with a plasma IL-33 concentration above the median than in those with lower levels (46.8% versus 32.7%; p=0.016).49
IL-33 is found in two forms in the body: IL-33RED and IL-33OX. IL-33RED is stored in the nuclei of structural cells such as epithelial and endothelial cells, and is rapidly released upon tissue injury and cell damage induced by trauma, infections, pollutants, and allergens, for example.46,50,51 IL-33RED undergoes a conformational switch to IL-33OX upon exposure to the extracellular environment.51
IL-33RED binds to the serum-stimulated-2 (ST2) receptor on immune and endothelial cells, which recruits IL-1 receptor accessory protein (IL-1RAP) to form a heterodimer that activates various inflammatory pathways, including Type 1 inflammation via cell types such as Th1 cells, Type 2 inflammation via cell types such as eosinophils, and Type 3 inflammation via cell types such as neutrophils and macrophages.45,50,52,53 IL-33 can also stimulate endothelial cells to release cytokines involved in Type 1 and Type 3 inflammation.54,55
IL-33RED is a potent inflammatory cytokine, and multiple homeostatic mechanisms regulate its activity. Firstly, IL-33RED is retained in the nuclei of airway epithelial cells and is inactivated by caspase 3/7 during apoptosis to prevent initiation of an immune response.56 Secondly, ST2 is found not only as a membrane-bound form but also as a soluble form (sST2) that acts as a ‘decoy’ receptor to dampen the immune response to IL-33RED.56 sST2 acts as an endogenous regulator of inflammation, and it is thought that reduced sST2 levels may cause an imbalance between IL-33RED and sST2 that promotes uncontrolled inflammation.57-59 Thirdly, the conformational switch that occurs on oxidation of IL-33RED to IL-33OX prevents it from binding to membrane-bound ST2 receptors.46,51
In vitro experiments have shown that IL-33OX signals via the receptor for advanced glycation end-products (RAGE)/epidermal growth factor receptor (EGFR) and is involved in mucus hypersecretion and airway remodelling.46 Experiments using human bronchial epithelial cells cultured in an air-liquid interface revealed that goblet cell MUC5AC/B expression was upregulated by IL-33OX, inducing a human epithelial mucin hypersecretion phenotype similar to that observed in COPD.46 MUC5AC secretion was also increased by IL-33OX but not by an oxidation-resistant form of IL-33RED (p≤0.01).46 The effects of IL-33OX and IL-33RED were also evaluated in a model of airway epithelial wound healing, which measured the extent of wound closure 24 hours after a scratch injury to cultured primary human bronchial epithelial cells. IL-33OX, but not IL-33RED, inhibited wound closure, indicating that IL-33OX impairs epithelial repair mechanisms.46
IL-33 dysregulation is a key driver of COPD pathogenesis, with IL-33RED-mediated pathways causing inflammation, and IL-33OX-mediated signalling leading to mucus hypersecretion and impaired epithelial repair (Figure 2).44,46,50
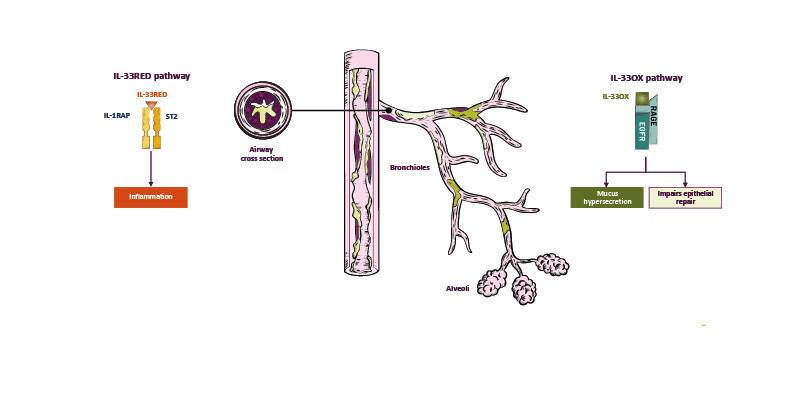
Figure 2: IL-33 dysregulation is a key driver of inflammation and mucus dysfunction in COPD.44,46,50,60
EGFR: epidermal growth factor receptor; IL-33OX: oxidised IL-33; IL-1RAP: IL-1 receptor accessory protein; IL-33RED: reduced IL-33; RAGE: receptor for advanced glycation end-products; ST2: serum-stimulated-2.
Smoking status has complex effects on IL-33 signalling. A recent study reported significantly higher sputum IL-33 levels for 80 people with COPD than for 20 healthy controls (median [interquartile range]: 38.7 [16–80] versus 14.1 [8–37] pg/mL; p<0.05).61 Interestingly, among patients with COPD, active smokers had significantly lower sputum IL-33 concentrations than former smokers (median [interquartile range]: 23 [11–53] versus 64 [32–108] pg/mL; p=0.002).61 Nonetheless, patients with severe COPD (GOLD Stage III/IV) had higher airway IL-33 levels compared to healthy controls, irrespective of their smoking status.61
An analysis of the association between smoking status and IL-33 gene expression across eight different studies also provided evidence of lower IL-33 gene expression in current smokers with COPD than in former smokers with COPD.62 However, there also appeared to be a trend toward lower ST2 gene expression in active smokers than in former smokers.62 The impact of this interplay between IL-33 and ST2 gene expression levels needs further elucidation, particularly given that ST2 is expressed not only as a membrane-bound receptor that mediates IL-33 signalling but also as a soluble protein that can lower the activity of this pathway. Notably, further analyses of gene set variation in bronchial epithelial air-liquid interface cultures have indicated that IL-33OX signalling is higher in patients with COPD than in healthy controls, and higher in current smokers than in former smokers, irrespective of COPD status.46 These data raise the possibility that active smoking may be associated with enhanced activation of IL-33OX signalling relative to former smokers.
Given its role as a key orchestrator of the inflammatory cascade (Figure 3), IL-33 is a strategic therapeutic target for COPD.45,46,50,51,63,64 Several novel biologics have been developed that inhibit IL-33 activity with differing mechanisms of action. Phase II and Phase III studies of these biologics are either completed or ongoing, and the results of these studies will provide important insights into the potential of these agents as novel COPD therapies.
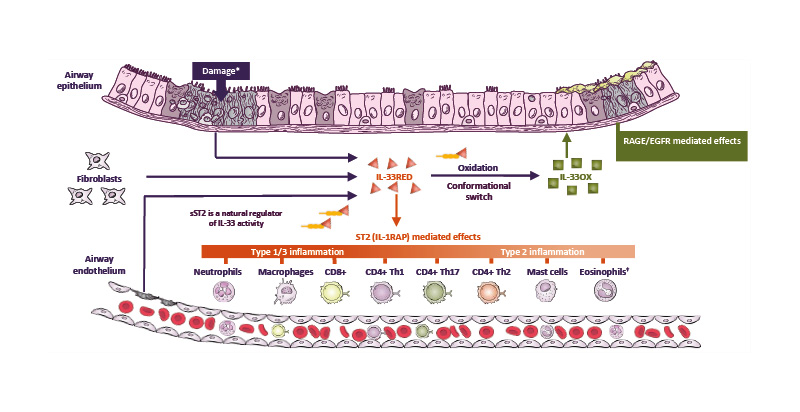
Figure 3: IL-33 is a key orchestrator of the inflammatory cascade in COPD and a strategic therapeutic target.45,46,50,51,63-66
*Damage induced by smoke, pollutants, and viral or bacterial exposure.
†Eosinophils are elevated in 10–40% of patients with COPD.45
CD4+/8+: cluster of differentiation 4/8-positive; EGFR: epidermal growth factor receptor; IL-33OX: oxidised IL-33;
IL-1RAP: IL-1 receptor accessory protein; IL-33RED: reduced IL-33; RAGE: receptor for advanced glycation end-
products; sST2: soluble serum-stimulated-2; ST2: serum-stimulated-2; Th1/2/17: T helper 1/2/17 cell.
Exploring Clinical Development of IL-33-Targeted Biologics
Stephanie Christenson
Phase II and Phase III clinical trials have been undertaken to evaluate the different types of biologics targeting IL-33 pathways in the management of COPD.67-83 Christenson began by summarising the clinical development programmes for the three types of biologics described above by Singh. They went on to present data from some of the Phase II studies that have been published.67,68,72,77,82,83 Although the primary endpoint was not met in the Phase II clinical trials for these biologics, important signals of clinical efficacy were observed, supporting the initiation of large Phase IIb/III programmes. The results of these Phase IIb/III studies have yet to be fully published, so Christenson focused on describing the important features of the design of each of these clinical trials.69,70,73,74,78-80 The results from the Phase IIb/III studies will help to better understand these molecules and elucidate the impact of their different mechanisms of action.
Panel Discussion
D’Cruz described the importance of treating both inflammation and mucus dysfunction in COPD. Although therapies are available to break down mucus and promote its expectoration, pharmacological interventions inhibiting mucus overproduction are lacking. Given the adverse effects of long-term OCS use,20 D’Cruz was of the opinion that new treatments are needed to reduce the risk of further exacerbations and steroid exposure in patients with COPD who exhibit exacerbations on triple therapy.
Singh discussed the COPDGene study35 and the fact that mucus plugs are associated with higher exacerbation and mortality rates.29,34 Establishing whether interventions that reduce mucus plugging improve outcomes was highlighted as important. Additionally, Singh addressed potential biomarkers for responders to anti-IL-33 therapy, highlighting exacerbation frequency as a disease severity measure that might help identify patients requiring more aggressive therapy.
Christenson explained that IL-33 is a good therapeutic target for COPD because it is involved in driving heterogeneous types of inflammation as well as mucus dysfunction, all of which are relevant to COPD. Discovering whether differences in mechanisms between biologics are reflected by differences in clinical trial outcomes will be enlightening. Christenson also stressed the importance of acquiring data for both current and former smokers in clinical trials.
Conclusion
Vogelmeier emphasised the unmet need for novel treatments targeting the mechanisms underlying COPD, including inflammation and mucus dysfunction. IL-33 is a potential therapeutic target for COPD because it promotes all these pathogenetic mechanisms through IL-33RED and IL-33OX. Novel biologics inhibiting the IL-33 pathway have differing mechanisms of action, and it will be interesting to establish whether these differences are reflected in the outcomes of Phase III trials.
Z4-77844 / November 2025






