INTRODUCTION
Primary immunodeficiencies (PID) are a heterogeneous group of genetic disorders that render the affected child susceptible to life-threatening infections, due to the absence or defect of one or more components of the immune system.
The diagnosis of PID is not only a complex process for a physician but also fills the lives of families of children with PID with dilemmas and queries. There is a dire need to raise awareness among physicians to streamline and accelerate the diagnostic process, help prevent end-organ damage, and provide definitive treatments, such as bone marrow transplantation (BMT) or other interventions.
So far, 485 distinct genetic defects have been identified, owing to advancements in genetic analysis.1 However, these, along with basic diagnostic workups, are not easily accessible due to high costs and a lack of physician awareness.1
For physicians performing workups for PID, it has become imperative to use resources cautiously, judiciously, and wisely, particularly in resource-limited countries. A methodical approach should be adopted, incorporating a detailed history, clinical evaluation, and laboratory investigations to elucidate and identify the deficient component of the immune system, even when diagnostic facilities are scarce.
CLASSIFICATION SYSTEMS
At a global level, two distinct systems are employed for the diagnosis and classification of PID. One is the International Union of Immunological Societies (IUIS) classification system devised by the IUIS Expert Committee. According to this, PID is now regarded as inborn errors of immunity (IEI), as there is not only a higher propensity of recurrent infections, but also autoimmunity, autoinflammatory conditions, allergic manifestations, and malignancies.1 In this classification, genotypic criteria complement the phenotypic criteria, ultimately categorising over 485 gene defects into 10 groups. Diagnosis is based on clinical features, immunological laboratory findings, and genetic confirmation where available.
The following are the 10 major categories of IEIs:
- Immunodeficiencies affecting cellular and humoral immunity.
- Combined immunodeficiencies with associated or syndromic features.
- Predominantly antibody deficiencies.
- Diseases of immune dysregulation.
- Congenital defects of phagocyte number or function.
- Defects in intrinsic and innate immunity.
- Autoinflammatory disorders.
- Complement deficiencies.
- Bone marrow failure syndromes.
- Phenocopies of IEI (acquired defects that mimic IEIs).
Another classification, by the European Society for Immunodeficiencies (ESID), provides practical diagnostic criteria for clinical and research use, especially when genetic confirmation is not available.2
The ESID defines PID diagnosis at three levels for each disease:
- Definite PID
The patient can be declared a definite PID case if a pathogenic mutation is found in a known PID-causing gene, or if the patient has a very specific clinical picture and family history along with immunophenotyping. - Probable PID
When there are strong clinical and immunological features suggesting PID, but no genetic confirmation is available. - Possible PID
When there is limited clinical or immunological data and further work-up is needed.
Depending on the specific clinical scenario and available facilities, one can adopt a system that is feasible, with a clear preference for the use of genetic testing for precise delineation.
USE OF SCREENING TOOLS IN SUSPECTED PATIENTS
Presentation of PID is often complex as the primary hallmark i.e., recurrent infections can be seen in other conditions, e.g., repeated infections in malnourished children living in unhygienic and poor sanitary conditions is specifically significant in developing countries. Thus, screening tools such as the warning signs published by JMF can be a useful guide for initial scrutiny.3 These include:
- four or more new ear infections within 1 year;
- two or more pneumonias within 1 year;
- two or more serious sinus infections within 1 year;
- two or more months on antibiotics with little effect;
- failure of an infant to gain weight or grow normally;
- recurrent deep skin or organ abscesses;
- persistent thrush in mouth or fungal infection on skin;
- need for IV antibiotics to clear infections;
- two or more deep-seated infections (e.g., sepsis, osteomyelitis); and
- family history of primary immunodeficiency.
In addition to these factors, the author has observed that recurrent and persistent diarrhoea is an important presenting complaint in T cell defects and severe combined immunodeficiency.
SALIENT FEATURES OF HISTORY AND EXAMINATION IN PRIMARY IMMUNODEFICIENCY EVALUATION
It is imperative to navigate the important points in history and examination to gather the basic information about patients with PID.
While evaluating history, the most important component is a detailed account of infection including age at symptom onset; site or location of infection; type of infection; and the frequency, severity, and seriousness of illness.4 The latter can be gauged by asking whether the patient only required outpatient visits or needed hospital and ICU admissions, and how the child responded to therapy.4
Over years of encountering such patients, it has been found that diagnostic delay should be evaluated by asking the age of symptom onset and the age of final diagnosis. This has multiple implications for the evolution of knowledge about PID. By evaluating the difference in diagnostic delay in specific types of PID documented in various studies at different points in time, one can determine not only the awareness level of physicians but also the effectiveness of diagnostic techniques like genetic testing. Besides determining the level of awareness among physicians and the general public, it enables the researcher to assess the nature and type of the underlying disease.
It is essential to ask the parents if they can tell or show any previously available reports indicating microorganisms that have grown in the past, which can potentially signal the deficient component of the immune system. This question should be asked explicitly, as parents are frequently not aware of the significance of such information and may forget to mention it when the history is taken.
It is of utmost importance that we try to find out the type of organisms by taking appropriate samples for identification, culture, and sensitivity. This gives a very strong indication regarding the deficient component of the immune system. For example, recurrent meningitis may raise the possibility of complement deficiencies.
Complement deficiencies and complement regulatory protein deficiencies are rare but important causes of immunodeficiency. A history of recurrent infections, particularly with encapsulated bacteria such as Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria species, must be noted, manifesting as sinusitis, otitis media, pneumonia, or severe conditions such as meningitis and sepsis.5 Recurrent sinopulmonary infections and autoimmune diseases involving classical pathway deficiencies, and repeated Neisseria infection involving both alternate and terminal pathway deficiencies are significant features.5
Past medical history, including delayed shedding of primary teeth, recurrent fractures, previous surgery (incision and drainage/biopsy), delayed separation of umbilical cord, previous hospitalisation, the number of previous admissions in hospital, and the duration of hospital stays, should be documented.
It is crucial to obtain detailed vaccination history, as live vaccines in patients with PID are dangerous and even life-threatening, depending on the type and severity of the immune defect.6 Document whether Bacillus Calmette-Guérin (BCG) was given, the use of polio and rotavirus drops, and any adverse events following vaccination.
A detailed family history needs to be elaborated, including early infant deaths due to infections, history of consanguinity, affected siblings with similar illnesses, and any other family members affected.6
It is important to recognise that autoimmune cytopenias like immune thrombocytopenia (ITP) and autoimmune haemolytic anaemia may be the initial or sole manifestation of common variable immunodeficiency, with one study reporting occurrences in 17.8% of cases.7 This can complicate the clinical picture and lead to diagnostic delays.
Patients may present with symptoms such as unexplained bruising, fatigue, or pallor, which are indicative of thrombocytopenia or haemolytic anaemia.
Assess for environmental or occupational exposures that may predispose patients to infections.6 Careful history-taking is required to rule out the possibility of recurrent chest infections due to anatomical defects or congenital conditions (e.g., repeated infections in congenital heart defects like ventricular septal defect, due to left ventricular volume overload; recurrent chest infections in cystic fibrosis and primary ciliary dyskinesia). A detailed history of early neonatal events can give clues for such diagnoses.
Recurrent chest infections are very common in patients with cerebral palsy, highlighting the significance of birth and developmental history (any history of delayed cry, and delayed achievement of milestones). A history of gastroesophageal reflux disease is equally crucial to discern.
It is also necessary to rule out any possibility of secondary immunodeficiency by taking detailed account of caloric intake for malnutrition assessment, any history of blood transfusions, and parental occupation (to rule out HIV infection). Features of protein-losing enteropathy or nephrosis which can lead to hypogammaglobulinaemia, should be inquired about. A history of drug intake, such as steroids or rituximab, must also be incorporated.
A detailed history should be followed by a complete physical examination. Foremost importance should be given to documenting signs of chronic ill health, such as pallor and failure to thrive. Perform anthropometric measurements and plot on centile charts; and if possible, ask for previous growth charts to compare with the current status.
Next, examine for signs of active infections like fever, cough, or lymphadenopathy. Specifically check for dysmorphic features, as they are distinctive for certain defects.8 For example, the following are classically observed in DiGeorge syndrome (DS): underdeveloped chin due to mandibular hypoplasia, low-set or posteriorly rotated ear, wide-set eyes, hypertelorism, antimongoloid slant to the eyes, and a short philtrum of the upper lip.
In hyper IgE syndrome, we may find prominent forehead, deep-set, widely-spaced eyes, a broad nasal bridge, a wide, fleshy nasal tip, mild prognathism, and facial asymmetry. However, it is important to note that these features may not be present at birth, but the patient develops these over the years due to multiple skin and other infections.
Clinical features of utmost importance include silver hair or hypopigmented hair, and partial albinism, seen in Chediak-Higashi syndrome. Ocular Telangiectasia seen in ataxia telangiectasia; hearing loss (due to repeated infections); absence of lymphoid tissue, tonsils, and lymph nodes (severe combined immunodeficiency disease and Bruton’s disease); gingivitis (phagocytic defect); oral thrush (seen in severe combined immunodeficiency disease, T cell defect, and phagocytic defects); and presence of clubbing (bronchiectasis). Moreover, a detailed skin evaluation should be conducted to discern rash and petechiae (Wiskott-Aldrich syndrome), evidence of scar tissue, abscess or granuloma formation, candidal infections, and any allergic manifestations. Additional features like lymphadenopathy and hepatosplenomegaly (suggestive of phagocytic defect or autoimmune lymphoproliferative syndrome), pallor, splenomegaly, bruises (indicative of autoimmune haemolytic anaemia and immune thrombocytopenia),7 gait disturbances (i.e., ataxia in ataxia telangiectasia, and Chediak-Higashi syndrome) should be noted.9 The physician must conduct a detailed examination of the primarily affected system, along with a systemic overview. Signs for immune dysregulation, like joint swellings, rashes, and alopecia, should also be noted.
DIAGNOSTIC WORKUP
In light of the information gathered, the physician can plan a list of investigations (Table 1), including complete blood count with neutrophils counts (neutropenia). It is imperative to determine absolute lymphocyte count and absolute neutrophil count; platelet number, size, and morphology (especially for Wiskott-Aldrich syndrome); reticulocyte count (autoimmune haemolytic anaemia); erythrocyte sedimentation rate (chronic Infection); and HIV screening (to rule out secondary Immunodeficiency).10
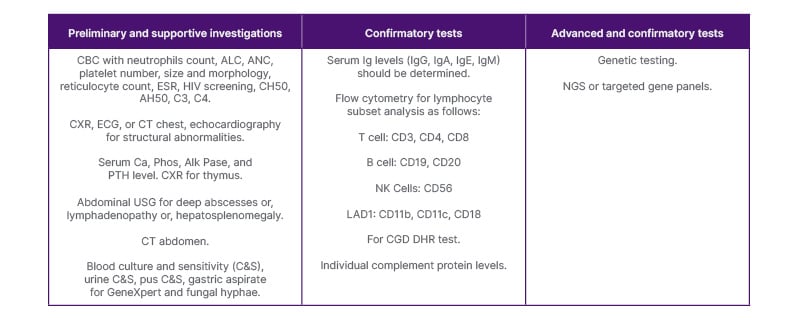
Table 1: Tiered investigations for primary immunodeficiency disorders.
AH50: alternative haemolytic complement; ALC: absolute lymphocyte count; Alk Pase: alkaline phosphatase; ANC: absolute neutrophil count; C&S: culture and sensitivity; Ca: calcium; CBC: complete blood count; C3: complement component 3; C4: complement component 4; CD3: cluster of differentiation 3; CD4: cluster of differentiation 4; CD8: cluster of differentiation 8; CD11b: cluster of differentiation 11b; CD11c: cluster of differentiation 11c; CD18: cluster of differentiation 18; CD19: cluster of differentiation 19; CD20: cluster of differentiation 20; CD56: cluster of differentiation 56; CGD: chronic granulomatous disease; CH50: total haemolytic complement activity; CXR: chest X-ray; DHR: dihydrorhodamine; ESR: erythrocyte sedimentation rate; LAD1: leukocyte adhesion deficiency Type 1; NGS: next generation sequencing; NK: natural killer; Phos: phosphate; PTH: parathyroid hormone; USG: ultrasound scan.
Serum immunoglobulin levels including IgG, IgA, IgE, and IgM should be determined. Flow cytometry should be conducted for lymphocyte subset analysis as follows:
- T cells: cluster of differentiation (CD) – CD3, CD4, CD8
- B cells: CD19, CD20
- Natural killer cells: CD56
- Leukocyte adhesion deficiency Type 1: CD11b, CD11c, CD18
In case there is a suspicion of chronic granulomatous disease and find hypergammaglobulinaemia, the dihydrorhodamine test should be done by flow cytometry. For screening of complement deficiencies, CH50, AH50, C3, and C4 can be done with further confirmation through individual complement protein levels. Advanced testing includes enzyme assays, such as adenosine deaminase (ADA). Genetic testing should be utilised when available with next-generation sequencing or targeted gene panels to identify the pathogenic mutations in PID. This is most helpful for precise delineation of the underlying mechanism. Such precise diagnosis can facilitate genetic counselling and prenatal diagnosis for future children.11
Supportive investigations include: chest X-ray, ECG, CT chest, and echocardiography for structural abnormalities; serum calcium, phosphate, alkaline phosphatase, parathyroid hormone level (DS); α-fetoprotein; and chest X-ray for thymus (lateral and frontal projection; T cell, severe combined immunodeficiency disease, DS).Abdominal ultrasound to document deep abscesses, or lymphadenopathy, or hepatosplenomegaly. If required, CT abdomen can be planned (chronic granulomatous disease).
One should aim to identify the offending microorganism from the appropriate sample depending upon the site of infection, e.g., blood culture and sensitivity (C&S), urine C&S, pus C&S, gastric aspirate for GeneXpert and fungal hyphae. Tests such as 50% haemolytic complement assay, antibody response to vaccines, and functional assays (e.g., lymphocyte proliferation assay) are practically not easily available.
CONCLUSION
Although approaching patients with PID is a cumbersome and complex process, a systematic approach incorporating targeted history, thorough examination, and appropriate investigation can streamline the process to not only eliminate the diagnostic burden on the patient but also to improve patient outcomes by enabling timely treatment. By delineating the specific defect and guiding treatment decisions, genetic testing has been a game-changer in this process.
There is a grave and pressing need to establish multinational and specialised centres, especially in developing countries, with the contribution of international bodies, where sustained availability of diagnostic facilities, including genetic testing, along with treatment facilities like Ig and haematopoietic stem cell transplantation is ensured.
The lack of such facilities in resource-constrained countries is evidence that global collaboration is the only solution to facilitate patients with PID, for whom not only is the diagnostic journey cumbersome and expensive, but the provision of Ig and preparation for bone marrow transplantation also impart a huge burden. One must not forget that these patients possess the right to live a normal and productive life. This right should not be stripped away on account of unaffordability.