
Abstract
Inflammasomes are sensors within the innate immune system that are responsible for the regulation of caspase-1 activation and the initiation of inflammatory responses following cellular infection or damage. A significant number of chronic inflammatory and metabolic diseases have recently been identified to have inflammasome-mediated inflammation as a key driver of their pathogenesis; this area of research is under intense investigation at present. This review focusses on autoinflammatory diseases (AD), a rapidly expanding group of debilitating diseases that are associated with severe systemic inflammation. AD commonly arise as a result of mutations to genes that encode inflammasome components. Monogenic AD are relatively rare because they require fully penetrating mutations; however, they often present at birth and last a lifetime. Clinical awareness of AD is lacking and it is believed that, at present, many cases go undiagnosed. This review specifically discusses a number of inflammasome-associated AD and metabolic disorders that provide significant insight into our understanding of inflammasome signalling pathways. These AD highlight the potency of inflammasomes in their ability to initiate and sustain systemic inflammation. The debilitating symptoms of AD also reveal the extensive consequences of uncontrolled inflammasome activity. Clinical therapies that target the inflammasome and interleukin-1β, a product of its activation, in the successful management of AD and certain metabolic diseases will also be discussed.
INTRODUCTION
The immune system is an evolutionarily conserved system that has evolved to protect the host from invading pathogens and cellular damage. While the immune system is crucial in protecting the host from a variety of insults, the dysregulation of immune components is strongly linked to the development of both autoinflammatory and autoimmune diseases. Autoinflammatory diseases (AD) are a relatively new category of immunological disease. The clinical term AD was proposed in 1999, when only two genes (MEFV and NLRP3) had been genetically associated with this disease category. Today, 30 genes have been linked to AD, which is the term still used to describe this expanding group of diseases, caused by the overactivation of the innate immune system. As this is a relatively new group of diseases, with new clinical subtypes being identified on an ongoing basis, there are limited statistical analyses available on AD. In 2013, a study1 estimated the incidence of AD to be 2.83 patients per million people in Sweden. Owing to their relatively recent identification and their low incidence rates, it is believed that clinical cases of AD are currently underdiagnosed and increased clinical awareness of AD is required.
Although both autoinflammatory and autoimmune diseases result from the immune system attacking the body’s own tissues, AD are characterised by intense episodes of inflammation, driven by innate immune cells, and are caused by mutations in genes that regulate innate immunity.2 The classical symptoms of autoinflammation are recurrent fever attacks, skin rash, and abdominal pain. However, AD symptoms vary greatly across clinical subtypes, and patients can present with a range of physical manifestations, including mouth ulcers, pyogenic skin or bone lesions, joint swelling, serositis, and granulomatous lesions.2 The number of gene mutations associated with AD is rapidly increasing. Examples of AD include cryopyrin-associated periodic syndrome (CAPS), Blau syndrome, familial mediterranean fever (FMF), tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), and inherited hyperimmunoglobulinaemia D with periodic fever syndrome (HIDS). The prevalence of a given disease can vary from 1:1,000 people (Sweet’s syndrome) to 1:1,000,000 (Marshall’s syndrome, also known as periodic fever adenitis pharyngitis aphthous [PFAPA] ulcers), and vary between populations. McGonagle and McDermott3 proposed a ‘continuum model’ for immunological diseases in 2006, integrating AD into a spectrum ranging from monogenic autoinflammatory disorders to monogenic autoimmune diseases, with polygenic autoinflammatory/autoimmune diseases and other diseases that may have both an autoinflammatory and an autoimmune component included within this spectrum. The majority of mutated genes identified to date linked to monogenic AD represent critical innate sensor or receptor proteins involved in inflammatory responses, such as NLRP3 for CAPS, NOD2 for Blau, and TNFR1 for TRAPS. Both NLRP3 and NOD2 proteins belong to the Nod-like Receptor (NLR) protein family, a group of cytosolic sensor proteins that are capable of detecting intracellular infection or damage. A number of the NLR proteins initiate inflammatory pathways by their formation of multiprotein complexes, termed inflammasomes. The majority of AD identified to date are in fact linked to mutations in inflammasome components.
INFLAMMASOMES: CRITICAL MEDIATORS OF INFLAMMATION
Inflammasomes are large complexes of proteins that form to mediate the activation of an inflammatory enzyme, termed caspase-1. Caspase-1 is transcribed as the inactive precursor protein pro-caspase-1, which requires proteolytic processing before the generation of its active form.4 Once active, caspase-1 is responsible for the maturation and secretion of interleukin (IL)-1β and IL-18, two potent proinflammatory cytokines that induce fever and interferon (IFN)γ secretion, respectively.5,6 In addition to the activation of the cytokines IL-1β and IL-18, inflammasome activation also results in a type of cell death, termed pyroptosis.7 Pyroptosis is an inflammatory form of cell death, mediated by caspase-1-dependent cleavage of an executioner protein, Gasdermin D (GSDMD).8 Cleaved GSDMD is responsible for forming pores in the cell membrane, mediating the release of proinflammatory cytokines IL-1β and IL-18.9 Thus, the ultimate outcome of inflammasome activation in cells is acute inflammation, driven by the secretion of potent inflammatory mediators IL-1β and IL-18, and pyroptotic cell death, which also contributes to local tissue inflammation in addition to eliminating damaged and infected cells.10
Inflammasomes are composed of a sensor protein, such as certain NLR proteins; an adaptor protein, usually the ASC protein; and the enzyme caspase-1. The sensor protein is responsible for recognising intracellular pathogens, such as bacteria or viruses, and/or intracellular danger or stress signals, such as detection of nuclear factors (e.g., DNA or high motility group box 1 protein [HMGB1]) in the cytosol. Once activated following the recognition of a pathogen or danger signal, the sensor protein oligomerises and triggers the formation of an inflammasome.11,12 Inflammasomes generally require a priming step before they can become activated, which is termed Signal 1. This priming step is mediated by NFκB signalling, which occurs following extracellular pathogen recognition by a toll-like receptor (TLR) or intracellular recognition by certain types of NLR, such as NOD1/2. Activation of NFκB during Signal 1 results in the transcriptional upregulation of inflammasome components, such as NLRP3 and pro-IL-1β, the inactive precursor form of IL-1β. Signal 2 involves activation and formation of the inflammasome complex via ligand binding to a sensor protein (Figure 1). An alternative method of NLRP3 inflammasome activation, known as the noncanonical inflammasome, requires a Signal 3, mediated by inflammatory caspases-4/5 in humans (caspase-11 in mice).13 Caspase-4, 5, and 11 are responsible for direct recognition of intracellular lipopolysaccharides, which results in their subsequent cleavage and activation.13 In addition to regulation of the noncanonical inflammasome,14 active caspases-4, 5, and 11 can also initiate pyroptosis, because they are capable of directly processing GSDMD.9 Several inflammasome complexes have been identified to date, including those that consist of NLR sensor proteins (NLRP1, NLRP3, NLRC4, NLRP6, and NLRP12) and other sensors, such as AIM2 and IFI16, which are members of the PYHIN protein family.15
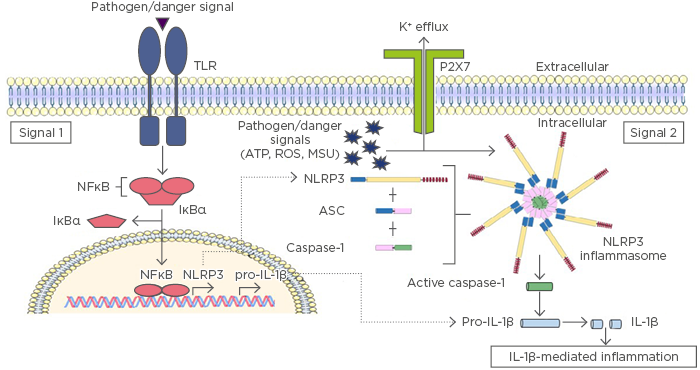
Figure 1: Inflammasome activation results in IL-1β mediated inflammation.
The initial priming step of inflammasome activation is mediated by pathogen recognition receptors, such as TLR, which recognise pathogen or danger signals during infection or injury. TLR activation results in translocation of NFκB into the nucleus to promote transcription and translation of inflammasome components and their targets, including IL-1β and NLR proteins (Signal 1). There are a number of mechanisms proposed to mediate Signal 2 activation; however, most occur via potassium (K+) efflux induced by pathogen or danger signals (e.g., ATP, ROS, MSU) and subsequent activation of NLRP3. NLRP3 oligomerisation initiates the formation of a multimeric inflammasome complex. The inflammasome complex facilitates the proteolytic cleavage and activation of caspase-1, allowing it to cleave pro-IL-1β into its mature and active form, which is secreted from the cell to mediate inflammation.
ATP: adenosine triphosphate; IL: interleukin; IκBα: inhibitor of NFκB; MSU: monosodium urate; NFκB: nuclear factor kappa B; NLRP3: NACHT, LRR, and PYD domains-containing protein 3; ROS: reactive oxygen species; TLR: toll-like receptor.
There have been >23 distinct NLR genes identified in the human genome, several of which have been implicated in the regulation and activation of inflammasome complexes, which subsequently lead to the activation and secretion of the proinflammatory cytokines IL-1β and IL-18. The most widely studied and best characterised of all inflammasomes is that of NLRP3. Extensive research has elucidated a range of microbial and nonmicrobial activators of the NLRP3 inflammasome. NLRP3 has been implicated in detecting a plethora of microbial pathogens, including the Influenza A virus, Vesicular stomatitis virus, bacterial Staphylococcus aureus, Escherichia coli, fungal Candida albicans, Aspergillus fumigatus, and parasitic Schistosoma mansoni and Dermatophagoides pteronyssinus.16-22 Additionally, phagocytosis of particulates, such as monosodium urate (MSU) crystals, amyloid-β, silica, calcium pyrophosphate dehydrate, asbestos, and alum have all been reported to activate NLRP3.23-27 These nonmicrobial agonists induce potassium efflux that results in subsequent NLRP3 activation. Recent studies28,29 have reported NEK7 as a novel NLRP3 inflammasome regulator. NEK7, a member of the NIMA-related kinase family, was originally found to be responsible for regulating mitotic progression and response to DNA damage but has since been reported to control NLRP3 oligomerisation, formation of an ASC speck, and subsequent caspase-1 activation downstream of potassium efflux and reactive oxygen species (ROS).28,29 Following their phagocytosis by innate immune cells, intracellular particulates are thought to damage the lysosomal membrane, resulting in the release of the lysosomal enzyme, Cathepsin B, into the cytosol, resulting in NLRP3 activation.30
INFLAMMASOME-MEDIATED AUTOINFLAMMATORY DISEASES
While inflammasome activation is a key mechanism responsible for mediating the host innate response following infection and injury, inappropriate inflammasome activity can lead to AD. As outlined previously, some of the well-characterised AD occur as a result of mutations in inflammasome-associated genes.
Mutations in NLRP3 have been linked to a group of disorders collectively known as CAPS, including familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disorder (also known as chronic infantile neurologic, cutaneous, and articular syndrome).31-33 All three CAPS subphenotypes occur as a result of dominantly inherited gain-of-function mutations in the NLRP3 gene, which result in systemic inflammation with blood neutrophilia and fever.32 Localised neutrophilic inflammation is also observed in various tissues, such as skin, muscles, joints, and cerebrospinal fluid. Symptoms common to all CAPS patients are rash, periodic fevers, headaches, joint pain, conjunctivitis, and general malaise. FCAS is the least severe of the CAPS and symptoms, which occur from early infancy, are triggered within 2 hours after cold exposure and generally subside within 24 hours.34 FCAS is distinct from cold urticaria, which is caused by an allergic response to cold and generally develops later in life. Symptoms in Muckle–Wells syndrome patients, triggered by cold, stress, or other unknown factors, are similar to those of FCAS but may also be accompanied by progressive hearing loss and the development of amyloidosis, due to excessive serum amyloid production.34 Neonatal-onset multisystem inflammatory disorder has the highest degree of chronic inflammation of all CAPS, with symptoms including aseptic meningitis, papilloedema, joint problems, hearing loss, and often mental and physical developmental delays.
Both in vitro and in vivo data support the hypothesis that CAPS-associated NLRP3 mutations result in enhanced responsiveness of the NLRP3 sensor protein, leading to inappropriate inflammasome activation and subsequent secretion of the potent inflammatory mediators, IL-1β and IL-18.35-38 Downstream markers of inflammation, such as IL-6, are also consistently elevated in patients with FCAS after a mild cold even when significant increases in IL-1β and IL-18 are undetectable.39 Additionally, pretreatment with anti-IL-1 therapy can prevent the FCAS response to a mild cold, suggesting a causative role for IL-1β in mediating the response.39 Approved and effective treatment options for CAPS patients now exist, as blocking the action of IL-1β using anakinra, rilonacept, or canakinumab are effective therapies for all CAPS patients.40 Therefore, the prognosis for all CAPS patients is greatly improved if the AD is diagnosed early and treated with the appropriate therapy before the damage caused by chronic inflammation has any permanent effect on the body. Mutations in the LPIN2 gene, encoding the lipin-2 protein, result in another NLRP3-associated AD, termed Majeed syndrome. Lipin-2 has been shown to regulate both the priming and activation steps of the NLRP3 inflammasome, and Majeed-associated LPIN2 mutations result in elevated pro-IL-1β and enhanced potassium efflux in macrophages, leading to aberrant NLRP3 activation.41
The most common AD is FMF, which occurs as a result of mutations in the MEFV gene, encoding the pyrin protein. Patients with FMF display longer periods of fever and can have a range of other symptoms, including skin rash, arthritis, and serositis.42 As the name suggests, FMF affects populations of Mediterranean descent, particularly Armenian, Turkish, Arabic, and some Jewish-Israeli populations, in which the carrier rates can be as high as 1:5.43-45 The high frequency carrier rates suggest that a selective advantage may exist, and previous reports have suggested that the mutated pyrin protein could provide increased protection against infection, asthma, or allergy.46-49
The pyrin protein, named due to the presence of a PYRIN domain in its protein structure, is thought to be responsible for protein–protein interactions. NLRP3 and other NLRP proteins are also characterised by the presence of a PYRIN domain, which is crucial for its ability to recruit ASC and other adaptor proteins into inflammasome complexes. In vitro pyrin overexpression studies reveal that, similar to NLRP3, pyrin oligomerises with ASC resulting in subsequent caspase-1 activation and release of IL-1β.50,51 To identify the impact of the MEFV Met694Val mutation, the most commonly found mutation in FMF patients, the Kastner group engineered a transgenic mouse strain that harboured the equivalent mutation in the murine Mefv gene.52 The genetically altered mice displayed FMF-like symptoms and also secreted high levels of IL-1β in an ASC-dependent manner, but Mefv deficient mice did not, suggesting that the FMF-associated mutations are gain-of-function, and cause enhanced and inappropriate inflammasome activation.52 Additional support for this hypothesis comes from a more recent study, which suggested that the pyrin inflammasome is negatively regulated by its phosphorylation of the pyrin protein, which is mediated by the RhoA signalling pathway.53 Under normal circumstances, the pyrin inflammasome is proposed to be selectively activated following RhoA GTPase inhibition by bacterial toxins;54 however, in FMF patients, mutated pyrin proteins are not efficiently phosphorylated by RhoA-dependent kinases, resulting in a lowered threshold for the activation of the pyrin inflammasome.53 This hypothesis is further supported by the fact that the antimitotic drug, colchicine, which inhibits microtubule polymerisation and activates RhoA, is an effective prophylactic treatment for FMF patients.
Inappropriate activation of the pyrin inflammasome is also linked to another unrelated AD: HIDS, also known as mevalonate kinase deficiency. HIDS is caused by mutations in the MVK gene, which encodes for the mevalonate kinase enzyme, an enzyme responsible for an early step in the isoprenoid synthesis pathway, catalysing the phosphorylation of mevalonic acid.55 RhoA signalling is dependent upon its translocation to the plasma membrane, which is regulated by the isoprenylation of RhoA.56 Defective isoprenoid synthesis occurs in the presence of mutations in the mevalonate kinase enzyme, resulting in loss of RhoA activity. Therefore, the molecular mechanism underlying the inflammatory symptoms of HIDS is also proposed to be mediated via the pyrin inflammasome.53 In contrast to FMF patients, colchicine is ineffective at preventing HIDS flares, most likely due to its inability to activate RhoA, which is not tethered to the membrane due to the absence of isoprenylation. Anti-IL-1β therapies are the main treatment option for HIDS patients, although not all patients respond. Other treatment options include nonsteroidal anti-inflammatory drugs (NSAID), glucocorticoids, and other biologics, such as TNF-α or IL-6 blocking agents.57
INFLAMMASOME ACTIVATION IN THE PATHOGENESIS OF METABOLIC DISEASE
The pathogenesis of many metabolic disorders, including atherosclerosis, Type 2 diabetes mellitus, obesity, and gout, is strongly associated with chronic inflammation. The inflammasome, and products of inflammasome activation (active IL-1β and IL-18), have recently been identified as key mediators of this inflammation, and thus are being intensively studied for their ability to modulate the pathogenesis and progression of metabolic disease. For example, results from the recent CANTOS trial58 reveal that targeting IL-1β-mediated inflammation reduces the risk of adverse cardiac events in patients with a previous history of myocardial infarction and high sensitivity C-reactive protein level (>2 mg/L). Additional analysis from the CANTOS study59 suggests that inhibition of IL-1β in these patients is also associated with reduced incidences of lung cancer. This suggests that further investigation into the use of anti-IL-1β and inflammasome targeting therapies for cancers with an established inflammatory component is warranted. However, the potential adverse effects that may arise when blocking such a potent inflammatory mediator must also be considered, as patients receiving canakinumab during the CANTOS trial had an increased occurrence of potentially fatal infections and sepsis.58
The contribution of the NLRP3 inflammasome to metabolic disease has been reviewed in great detail elsewhere;60,61 however, the proposed role of the inflammasome in the molecular pathogenesis of gout is summarised here as an example. Gout is a chronic inflammatory disease characterised by deposition of MSU crystals in joints, which form when high concentrations of urate are present. The clinical symptoms of gout arise as a result of the inflammatory response that occurs following recognition of the MSU crystals. Gout is believed to be the most common cause of inflammatory arthritis, with an increasing prevalence in both developing and developed countries.62 Activation of the NLRP3 inflammasome in gout has been well investigated and it is believed that TLR activation most likely acts as the first priming signal in the response to MSU crystals.63 Phagocytosis of MSU crystals by macrophages, which causes lysosomal damage and subsequent activation of the NLRP3 inflammasome constitutes Signal 2 (Figure 1). IL-1β has been implicated as a key inflammatory mediator responsible for driving the development of gout by promoting an influx of neutrophils into the synovium and joint fluid, which is a typical hallmark of an inflammatory bout in this disease.64 Anti-inflammatory therapies such as NSAID and glucocorticoids are effective in reducing the symptoms of gout. Colchicine is also prescribed as a prophylactic treatment or to relieve gouty flares. As described previously, colchicine inhibits microtubule polymerisation and, in contrast to the pyrin inflammasome, has been shown to disrupt NLRP3 inflammasome activation. Colchicine also inhibits microtubule-based inflammatory cell chemotaxis, secretion of chemokines and cytokines, and phagocytosis. Many of these cellular processes can be found in other diseases involving chronic inflammation, suggesting the potential efficacy of low-dose colchicine in other comorbid conditions associated with gout, such as osteoarthritis and cardiovascular disease.65
CONCLUSION
Anti-IL-1β and inflammasome targeting therapies are emerging as important clinical treatments for the management of AD, metabolic diseases, and certain cancers. Although inflammasome activation may not be the primary cause or major pathogenic factor for many metabolic diseases, recent evidence suggests that targeting the inflammatory contribution to these diseases may limit their progression. In contrast, certain monogenic AD, including CAPS, FMF, and HIDS, have been reported to arise directly as a result of defective and uncontrolled inflammasome activation. The fact that many AD are effectively treated by IL-1β blockade and drugs that target inflammasome activity highlights the potency of inflammasomes in driving chronic inflammation. As the mechanisms governing inflammasome regulation continue to evolve, so too will additional targets and therapies to regulate inflammasome activity during disease. However, the importance of controlled, functional inflammation for homeostasis cannot be ignored. Thus, therapeutic inflammasome inhibition needs to be balanced against the beneficial contribution of inflammasomes to innate immunity.





