
Abstract
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) represents a heterogeneous group of rare diseases characterised by necrotising inflammation of the small blood vessels and the presence of ANCA with specificity for proteinase-3 or myeloperoxidase. Genetic susceptibility along with malignancy, drug exposure, and environmental exposures to infectious agents and silica are involved in disease progression. To date, growing evidence has revealed that ANCA specificity defines homogeneous groups of patients more effectively than clinical diagnosis, since proteinase-3 ANCA and myeloperoxidase-ANCA are linked with different genetic backgrounds and epidemiologies. This review presents current and updated knowledge on the central aetiopathogenic role of genetic associations and environmental exposures in AAV; discusses the main mechanisms of ANCA immunogenesis; and highlights the value of ANCA specificity for future classification criteria.
INTRODUCTION
The anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV) are rare, potentially life-threatening, multisystemic autoimmune diseases, identified by the nomenclature of the 2012 revised International Chapel Hill Consensus Conference (CHCC) as a specific group of small vessel vasculitides different from the immune complex-mediated vasculitides.1 According to their clinical and pathological features, AAV have traditionally been subclassified into three clinicopathologic variants: granulomatosis with polyangiitis (GPA), formerly Wegener’s granulomatosis; microscopic polyangiitis (MPA); and eosinophilic granulomatosis with polyangiitis (EGPA), formerly Churg–Strauss syndrome.1 However, the traditional classification of AAV based on clinical phenotype has recently been challenged on epidemiological and genetic grounds. Accumulated evidence now suggests that ANCA specificity for the two major target antigens, myeloperoxidase (MPO) and proteinase-3 (PR3), could better define homogeneous groups of patients compared to those identified by clinical diagnosis.2,3
As with most autoimmune diseases, the aetiopathogenic process of AAV is likely to be multifactorial, varies among individuals, and involves a complex interplay between innate and acquired characteristics of the immune system, as well as environmental exposures and genetic influences. In this review, the authors summarise the central role of genetic associations and environmental exposures in AAV aetiopathogenesis and immunogenesis of ANCA, and underline how current epidemiological and genetic data could guide an improved classification system based on ANCA specificity.
EPIDEMIOLOGY AND GENETIC ASSOCIATIONS IN ANTI-NEUTROPHIL CYTOPLASMIC ANTIBODY- ASSOCIATED VASCULITIS
The annual incidence of AAV is approximately 13–20 cases per million individuals and the prevalence ranges from 46–184 cases per million individuals worldwide. EGPA is less common than GPA and MPA, with an annual incidence in the range of 0.5–2.0 cases per million individuals and a prevalence of 10–45 cases per million individuals.4,5 The sex distribution is similar and the peak incidence occurs in the middle of the sixth decade of life.4 Over the past few years, the increasingly well-documented epidemiology of AAV has highlighted geographical and ethnic differences according to clinical variants and their associations with ANCA subtypes. For example, GPA is more common in the north of Europe, while MPA is more common in southern Europe.6 The populations studied in western USA, Australia, and New Zealand were predominantly white Caucasians of northern European descent, and the incidence of GPA in these areas has been reported as very similar to Europe.5 A multiethnic population study conducted in Paris, France, reported that the prevalence of AAV in individuals of European descent (104.7 cases per million) was twice that of non-Europeans (52.5 cases per million). In the same study, GPA was less frequent in people of non-European ancestry than MPA, and none of the GPA patients came from Africa.7 In a multiethnic series from Chapel Hill in the USA, GPA was also relatively uncommon in African-Americans compared to Caucasians.8 Two geographical regions with a high incidence of MPA and much fewer GPA cases are Japan and Kuwait.9,10 A large case series also reported that MPA is more common than GPA in China.11
In European studies, the ANCA antigen specificity of GPA is most frequently associated with PR3, while the specificity of MPA is more commonly associated with MPO. However, the overlap between ANCA specificity and the clinical syndrome is only partial. PR3-ANCA cases are present in approximately two-thirds of patients with GPA, but also in a quarter of patients with MPA, whereas MPO-ANCA cases are present in the majority of patients with MPA, but also in up to a quarter of patients with GPA.12 In China, 60% of GPA patients have MPO-ANCA,13 while in Japan, MPO-ANCA is present in 83% of cases of AAV, including a significant proportion of those with GPA.9
Recent developments in the understanding of genetics could now explain these geographical and ethnic differences and help us to achieve a better understanding of the disease aetiopathogenesis. Over recent years, several studies, based on candidate gene approaches, have been performed in the field of AAV genetics. However, many of these studies were small and not replicated. Knowledge of AAV dramatically increased after the publication of two genome-wide association studies (GWAS) conducted by the European Vasculitis Genetic Consortium (EVGC)14 and the Vasculitis Clinical Research Consortium (VCRC).15
The EVGC GWAS was performed in a cohort of UK AAV patients and matched controls. This analysis reported both major histocompatibility complex and non-major histocompatibility complex associations with AAV. Subsequently, 158 single nucleotide polymorphisms (SNP) were genotyped in a replication cohort of northern European AAV patients and controls to confirm the GWAS data. Combined analysis of the two cohorts showed that the strongest genetic associations were with the antigenic specificity of ANCA (PR3 or MPO) rather than the clinical syndrome (GPA or MPA). HLA-DP; SERPINA1, which encodes alpha-1 antitrypsin, a serine protease that represents the major inhibitor of PR3 activity; and PRTN3, which encodes PR3, were in association with GPA and stronger association with PR3-AAV.14 Their specific loci and SNP were given as follows:
- HLA-DP (6p21.32, SNP rs3117242)
- PRTN3 (19p13.3, SNP rs62132295)
- SERPINA1 (14q32.13, SNP rs7151526)
A weaker association was found between MPO-AAV and HLA-DQ (6p21.32, SNP rs5000634).
The VCRC GWAS was conducted in a population of GPA patients and controls of European ancestry. The strongest association was with HLA-DPB1 (6p21.32, SNP rs9277554) and HLA-DPA1 (6p21.32, SNP rs9277341), and even more significant among the PR3-ANCA-positive subgroup. An independent SNP (rs26595) near SEMA6A on chromosome 5 was also associated with GPA, reaching genome-wide significance,15 but this was not confirmed in an independent cohort of European patients.16 The association between SERPINA1 and GPA was also confirmed in this GWAS.15
Interestingly, the HLA-DRB1*15 allele has been associated with PR3-AAV in African-Americans,8 whereas HLA-DRB1*1202 has been associated with PR3-AAV in a population of Han Chinese.17 Furthermore, it is now possible to speculate that the uncommon appearance of PR3-AAV in China and Japan is due to the low frequency of the HLA-DPB1*0401 and the PI*Z allele of SERPINA1 genetic variants.5
A recent meta-analysis performed by EVGC using 62 studies, including the 2 GWAS, identified 33 genetic variants in or near 15 genes associated with AAV:18 CD226 antigen, cytotoxic T lymphocyte-associated antigen-4, Fc fragment of IgG receptor IIa, HLA-B, HLA-DP, HLA-DQ, HLA-DR, hydroxysteroid 17-beta dehydrogenase 8, IFN regulatory factor 5, protein tyrosine phosphatase non-receptor type 22, ring finger protein 1, retinoid X receptor beta, STAT4, SERPINA1, and toll-like receptor 9. Moreover, genetic distinctions between GPA and MPO and between PR3-AAV and MPO-AAV were identified.18 These data provide clear evidence that GPA and MPA have distinct genetic associations and support the concept that PR3-AAV and MPO-AAV are separate autoimmune syndromes.
ENVIRONMENTAL ASSOCIATIONS AND IMMUNOGENICITY OF ANTI-NEUTROPHIL CYTOPLASMIC ANTIBODIES
Infections
The seasonal or temporal oscillations of AAV incidence reported in various epidemiological studies19,20 could be suggestive of an infectious agent. Recently, Draibe et al.21 confirmed, in Catalonia, Spain, the seasonal periodicity of AAV, showing it had a higher incidence in the winter.
Particular attention has been given to Staphylococcus aureus since chronic nasal carriage of S. aureus correlates with relapsing disease in patients with GPA.22 Conversely, antimicrobial therapy with cotrimoxazole leads to a substantial reduction in disease relapse incidence.23 These findings are supported by the fact that the immunisation of rats with bacterial proteins derived from S. aureus has been shown to cause AAV.24 Other infectious agents have also been associated with the onset of AAV, especially in patients with subacute bacterial endocarditis. In an analysis of 50 AAV case reports, three species of bacteria (Streptococci, Staphylococci, and Enterococci) were associated with 63% of the reported episodes of infectious endocarditis and 42% of all infections reported.25 Huan et al.26 recently demonstrated that ANCA-positivity in patients with Mycobacterium tuberculosis infection is related to AAV, so the detection of ANCA in these patients must be excluded because GPA and MPA can present with clinical features that overlap with those of tuberculosis. Among the viruses suspected to play a pathogenic role in AAV, parvovirus B19,27 Epstein–Barr virus,28 and Ross River virus have been reported.29
The involvement of infectious agents in the pathogenesis of AAV could be explained through a process known as ‘neutrophil priming’. Infections induce the production of proinflammatory cytokines, such as TNF-α, IFN-γ, and IL-1, leaving neutrophils and monocytes in a pre-activated (‘primed’) state by the migration of autoantigen targets (PR3 and MPO) to the cell surface to be presented to and activated by ANCA.30
Infectious agents can also be involved in the immunogenicity of ANCA. The so-called ‘autoantigen complementarity theory’ argues that the initial immune reaction is developed against a complementary peptide (c-peptide) to the self-antigen (original autoantigen). The c-peptide could be produced from the transcription of an endogenous antisense DNA strand or could be a mimic of an antisense peptide that is produced by a pathogen. Therefore, the initial immune reaction against the c-peptide results in the formation of anti-c-peptide antibodies, which in turn produce anti-idiotypic antibodies that identify not only the idiotope on the anti-c-peptide antibodies, but also the epitope on the sense peptide (original autoantigen).31 Numerous microbial homologues of the complementary PR3, including antigens from microbes correlated with GPA and PR3-ANCA, such as S. aureus, Ross River virus, and Entamoeba histolytica, have been described.32
An alternative theory based on molecular mimicry between bacterial and self-antigens has been proposed as a mechanism for the immunogenesis of ANCA. Kain et al.33 reported the presence of ANCA against lysosomal membrane protein-2 (LAMP-2) in approximately 90% of patients with active ANCA associated with necrotising crescentic glomerulonephritis. More importantly, the researchers showed that human LAMP-2 has 100% homology with the bacterial adhesin FimH, which is found in Gram-negative bacteria such as Escherichia coli and Klebsiella pneumonia. Accordingly, rats immunised with FimH developed pauci-immune necrotising crescentic glomerulonephritis and antibodies to rat and human LAMP2.34 However, this finding was not confirmed by Roth et al.35 in another animal model using Wistar–Kyoto rats. Therefore, the pathogenicity of LAMP2 antibodies remains uncertain. Recently, in another experimental model of molecular mimicry, Kim et al.36 showed that immunisation of C57BL/6 mice with a recombinant serine protease of Saccharomonospora viridis, which has >30% homology with human PR3, produced high levels of ANCA against mouse PR3.
Since stimulation of toll-like receptor 9 by bacterial DNA is known to promote B cell proliferation and antibody production,37 Hurtado et al.38 investigated and confirmed the possibility that unmethylated CpG oligodeoxynucleotides, found in bacterial and viral DNA, stimulate circulating autoreactive B cells to produce ANCA in patients with AAV.
Silica
Silica exposure has been associated with several autoimmune disorders,39 and various case–control studies have documented that 22–60% of patients with AAV, principally those with MPO positivity, were previously exposed to silica.40,41 Hogan et al.41 showed that there is an enhanced risk for the disease with long lifetime silica exposure, but there is no evidence of a correlation between the length of time since last exposure and disease onset. It has been suggested that silica exposure could evoke accelerated apoptosis of polymorphonuclear leukocytes and macrophages. The released MPO could then be taken up by alveolar macrophages and presented to immunocompetent cells, resulting in the development of anti-MPO antibodies.42 Furthermore, in vitro studies have showed that silica can activate apoptosis in human peripheral blood lymphocytes, with an increase in Fas-mediated cell death.43 Fas expression was significantly higher in Treg for silicosis patients compared with healthy donors, and when cultivated with an agonistic anti-Fas antibody, Treg from silicosis patients also showed a higher degree of apoptosis.44 Consequently, the reduction of Treg function makes these patients more sensitive to a disruption of self-tolerance.
Drugs
Drug-induced AAV has also been described (Box 1). The most common drug implicated with AAV is propylthiouracil (PTU), described in 80–90% cases of AAV cases induced by antithyroid drugs.45 Evidence for an association with the development of drug-induced AAV has also been shown for several drugs: hydralazine, minocycline, levamisole-adulterated cocaine, TNF-α agents (adalimumab, etanercept, and infliximab), sulfasalazine, D-penicillamine, allopurinol, psychoactive agents (clozapine, thioridazine), cefotaxime, and phenytoin.46 However, in most cases, the evidence is restricted only to case reports and the causative link is much less convincing.
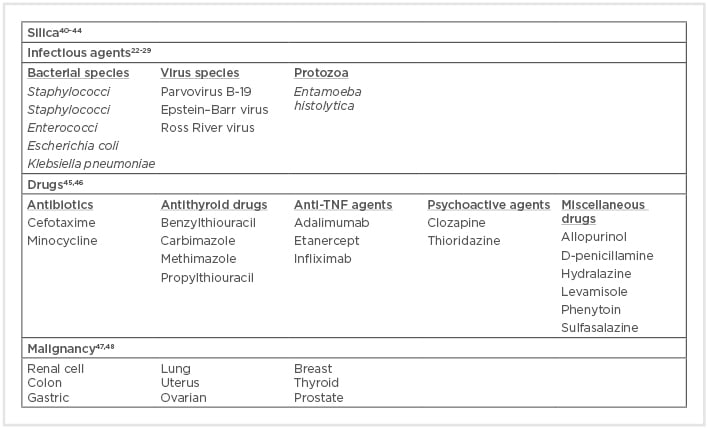
Box 1: Environmental associations with anti-neutrophil cytoplasmic antibody formation and anti-neutrophil cytoplasmic antibody-associated vasculitis.
PTU is used to treat hyperthyroidism due to its ability to inactivate thyroid peroxidase. Human MPO and thyroid peroxidase nucleotide sequences display an average similarity of 46%, which becomes 58% within the sequence encoding the functional MPO protein, while the similarity of the amino acid sequences is 44%.49 Thus, PTU treatment could reduce or competitively inactivate the oxidation activity of MPO in a dose-dependent manner, also altering its configuration.50 Therefore, the altered MPO might serve as a neoantigen. Jiang et al.51 proposed that drugs such as PTU and hydralazine in the presence of extracellular hydrogen peroxide could be transformed into cytotoxic metabolites by neutrophil-derived MPO. Under such conditions, the drugs and their metabolites are immunogenic for T cells, which in turn activate B cells to produce ANCA.52
Hydralazine, a DNA methylation inhibitor with suspected reverse epigenetic silencing properties, has been shown to increase the expression of MPO and PR3 autoantigens in neutrophils.46 Some drugs, such as sulfasalazine, could activate neutrophil apoptosis, which, in the absence of priming, is related to translocation of ANCA antigens to the cell surface and a subsequent production of ANCA.53
ANCA with specificity to human neutrophil elastase are rare and infrequently detected in patients with AAV; however, they might be useful for the diagnosis of cocaine-induced midline destructive disease and AAV associated with other drugs, such as PTU and hydralazine.46,54
Malignancy
Malignancy could be a complication in AAV because immunosuppressive therapy decreases the immune system’s ability to recognise and eliminate malignant cell clones and may have direct mutagenic properties.55 However, AAV occasionally develops secondary to malignancy. Paraneoplastic AAV is uncommon and accounts for <5% of all paraneoplastic vasculitis.56 Tatsis et al.47 reported 14 patients diagnosed with GPA and simultaneously occurring cancer. In a retrospective review of 200 patients with GPA or MPA, 20 patients had concurrent malignancy (diagnosed within 6 months), and solid tumours were most common, notably colon cancer, breast cancer, and gynaecological cancer.48 However, other studies have contradicted these results, reporting that the overall risk for malignancy prior to AAV diagnosis was similar to that of the general population.57,58
The exact pathophysiology of paraneoplastic-associated vasculitis is thought to be multifactorial.59 Postulated mechanisms include increased cellular turnover leading to generation of autoantibodies with the formation of immune complexes of tumour-associated antigens/antibodies that cannot be appropriately cleared; release of tumour angiogenic factors and/or cytokines, which induce endothelial damage and increase vascular permeability, inflammation, and fibrosis; and direct effects of tumour cells on vascular walls. The detection of PR3 in a human renal cancer cell line,60 in association with the demonstration that PR3 can act as a growth factor in non-haematopoietic cells, analogous to its role in haematopoietic cells,61 and the increased risk for renal cancer in GPA,47 suggest that a relationship could exist between PR3-positive renal cell cancer and the development of AAV.
Currently, further studies are needed to support the hypothesis that preceding malignancies and AAV have a causal relationship or shared pathogenic pathways. The microbial and non-microbial environmental factors, like silica and drugs, that have been linked to AAV are summarised in Box 1.
CONCLUSION
In this review, the authors document that, on the basis of recent genetic and epidemiological evidence, PR3-AAV and MPO-AAV are distinct autoimmune syndromes and, thus, new classification criteria for AAV are needed, which should include ANCA specificity as a major factor. The ongoing DCVAS study62 is a multinational observational study designed to develop diagnostic criteria and improve classification criteria for six forms of primary systemic vasculitis, including AAV. Furthermore, like in most autoimmune conditions, considerable progress has been made in our knowledge of the immunogenesis of AAV. The interaction between predisposing genes and triggering environmental factors is relevant also in AAV, leading to the loss of tolerance against self-antigens. These findings can now be tested in future clinical trials using new therapeutic strategies in well-defined homogenous patient populations according to their ANCA specificity.






