Abstract
Leukocyte adhesion deficiency type-1 (LAD-1) is a rare primary immunodeficiency that is characterised by compromised neutrophil adhesion and transmigration to infection or inflammation sites. In this article, the authors report the cases of five patients with LAD-1 deficiency. The aim of this study is the demonstration of the wide variety of manifestations in these patients with a review of the literature. Patients were both male and female, with their ages ranging from 1 month to 10 years old. Omphalitis was the most common presentation in this series, followed by pneumonia and gingivitis. Impaired wound healing and cellulitis were other common findings in these patients. Two of the patients did not show delayed separation of the umbilical cord. The findings indicate that LAD-1 remains a life-threatening condition with omphalitis, oral, skin, respiratory tract, and ear infections as the most common complications. Early identification of these patients is essential in ensuring a definitive diagnosis and early implementation of haematopoietic stem cell transplantation.
INTRODUCTION
Leukocyte adhesion deficiency type-1 (LAD-1) is a rare autosomal recessive primary immunodeficiency caused by mutations in the ITGB2 gene, which encodes the CD18 subunit of the β2 integrins, leading to compromised neutrophil adhesion and transmigration to infection or inflammation site.1,2 LAD-1 severity is related to the degree of CD18 deficiency.3 Leukocyte trafficking from circulation to tissues, as a critical response to integrins, serves as an essential dynamic process in immune surveillance and is mediated with a set of integrin molecules on leukocyte cell surfaces.4,5 Genetic alterations in the numbers or activation of leukocyte integrins may lead to the development of LAD-1 syndromes.6,7 Members of the β2 subclass of integrins are essential for normal adhesion-dependent functions of polymorphonuclear leukocytes and monocytes by providing normal integrin dimerisation.8-11 Deficient expression of each of the following four members of the β2 integrin subfamilies leads to LAD-1 syndrome: αLβ2(CD11a/CD18 and LFA-1), αMβ2 (CD11b/CD18, MAC-1, and CR3), αXβ2 (CD11c/CD18, and p150/95), and αDβ2 (CD11d/CD18). The β2 integrins are also expressed by other immune effector cells, including macrophages and lymphocyte subsets.12-15 αMβ2 and αLβ2 serve as the C3b receptor in myeloid and lymphoid cells16 and αDβ2 is restricted to macrophage subsets in mice and humans.12,17,18 The ITGB2 gene encoding for the β2-integrin component CD181,2,11,19 is located at the long arm of chromosome 21q22.3,3 and has >80 mutations identified so far. The severity of the clinical manifestations is directly related to the degree of CD18 deficiency,3 with the mild-to-moderate types characterised by 2–30% CD18-expressing neutrophils and severe LAD-1 classified as <2% CD18-expressing neutrophils.11,19,20
Patients with LAD-1 typically present with recurrent indolent necrotic infections of the skin and mucosal surfaces, which usually occur during the neonatal period with omphalitis and delayed separation of the umbilical cord.3 Additionally, failure to thrive, severe malnourishment, and delayed wound healing are common, while colitis may only occur in rare cases.21 Many patients may develop infections before they are diagnosed with LAD-1 because many physicians are unaware of the disease.22 Here, the authors describe five cases admitted to Mofid Children’s Hospital, Tehran, Iran, with different presentations and prognoses to emphasise the importance of disease awareness and timely diagnosis in the successful management of the disease.
METHODS
The patients were evaluated with a detailed history, physical examination, and immunologic screening tests at the time of their first admission to hospital. Complete blood counts (CBC) were performed using an automated blood counting machine (Sysmex XE-2100 [Sysmex, Kobe, Japan]). Haemagglutinins were quantified by CA 1600 and immunoglobulin levels were evaluated by ELISA. Review of CD markers was conducted by flowcytometry analysis (PAS system [Partec, Munich, Germany]).
The study was approved by the ethics committee of Mofid Children’s Hospital. The authors also established clear policies to secure patients’ privacy.
CASE 1
A 45-day-old full-term infant was admitted due to fever, poor feeding, and vomiting. She was first referred to the authors’ hospital at this age due to refractory sepsis that did not respond to antibiotics. The infant was a consanguineous product, born to a 28-year-old mother by normal vaginal delivery, and she presented with normal Apgar scores and appropriate weight for gestational age. At the time of admission, she was sick and febrile (39 oC). The umbilical cord had been separated but the parents reported a significant delay in its detachment (at 30 days). Following physical examination, periumbilical erythema with serous discharge, suggesting omphalitis, was detected. Laboratory investigations revealed significant leukocytosis (a leukocyte count of 59,900 mm3 with 78% polymorphonuclears). Immunophenotyping performed by flow cytometry analysis reported low CD18 (1.0%), CD11a (2.0%), CD11b (3.0%), and CD11c (0.9%) antigens, which is consistent with LAD. Haematopoietic stem cell transplantation (HSCT) has not been conducted because a matched donor has not yet been found. However, several follow-up visits have been conducted until the present day. She is currently aged 3 years.
CASE 2
A 40-day-old female infant, the only child of consanguineous parents, was admitted to hospital three times. Her third admission was due to fever, omphalitis without pus, and poor wound healing at the site of a previous intravenous line from her last hospital admission. Her past medical history revealed two previous admissions, with the former due to omphalitis and the latter due to soft palate lesions mimicking fungal infections. She had a history of delayed umbilical detachment (at 23 days of life). The laboratory tests at the time of admission revealed leukocytosis (80,700 mm3) with neutrophil predominance (72%) that persisted throughout the course of hospitalisation despite appropriate antibiotic therapy. Flow cytometry analysis detected low levels of CD11a (0.70%), CD11b (0.35%), CD11c (0.23%), and CD18 (0.10%). She had follow-up visits regularly for 9 years, but, because of the lack of a matched donor, she received HSCT at the age of 9 years at a late stage when she had nearly experienced the late sequela of the disease. She died 1 month after the transplantation due to infectious complications.
CASE 3
A 10-year-old girl, the product of a twin pregnancy from unrelated parents, was admitted to the authors’ hospital due to necrotising skin lesions on the left inguinal region 3 years ago at the age of 7 years. There were also multiple scarring lesions on both extremities due to recurrent cellulitis beginning from 3 years before admission. She has a history of both delayed umbilical cord detachment in her neonatal period and also delayed wound healing since infancy (at the age of 8 months). She had recurrent attacks of gingivitis. The laboratory tests revealed moderate leukocytosis (34,800 mm3) with neutrophil predominance (67.0%) at the time of admission. Flow cytometry analysis detected low levels of CD18 and CD11a (9.1% and 0.1%, respectively). A successful human leukocyte antigen-identical HSCT was performed from her healthy twin sibling last year. She has had several follow-up visits after HSCT for 1 year until recently and she is responding well.
CASE 4
In 2017, a 7-year-old boy, who was the only child of unrelated parents, presented with recurrent gingivitis, but responded well to oral antibiotics and mouthwash every time. His past medical history was indicative of omphalitis in the neonatal period. The umbilical cord separation was not delayed. His family history revealed that his cousin had LAD at an early age and died as a result. A CBC at the time of admission showed 30,700 white blood cells (WBC) with neutrophil predominance (71%). Flow cytometry analysis detected CD11a: 0.1%, CD11b: 92.0%, CD11c: 6.0%, and CD18: 0.1%. HSCT has not been performed for him. However, he is still alive and has been undertaking follow-up visits for the last 2 years.
CASE 5
A 2-year-old boy from consanguineous parents was admitted with a history of recurrent bilateral otitis media and mastoiditis from early infancy (age of 2 months). At the time of his first admission in our hospital at the age of 2, he presented with left leg cellulitis, which had not been responsive to several courses of oral antibiotic therapy. At 3 years old, he had a perianal abscess and has had recurrent buttock cellulitis since then. CBC revealed leukocytosis (WBC count: 28,000 mm3) with neutrophil predominance (75%). Flow cytometry analysis showed low numbers of CD11a (1.5%), CD11b (1.0%), CD11c (4.0%), and CD18 (2.0%). A matched donor was not available for him and he died because of pneumonia and fulminant sepsis from his last admission at another hospital. He had been participating in follow-up appointments for 6 years before his death at the age of 8 years.
RESULTS
Patients’ age, sex, clinical features, parental consanguinity, laboratory findings, treatment, and outcome are summarised in Table 1. Omphalitis was the most common presentation in this series, followed by pneumonia and gingivitis. Impaired wound healing and cellulitis were other common findings in these patients. One patient presented with fungal infection. Laboratory evaluation was indicative of severe LAD (according to CD18 <2% in flow cytometry analysis) in four of the patients.
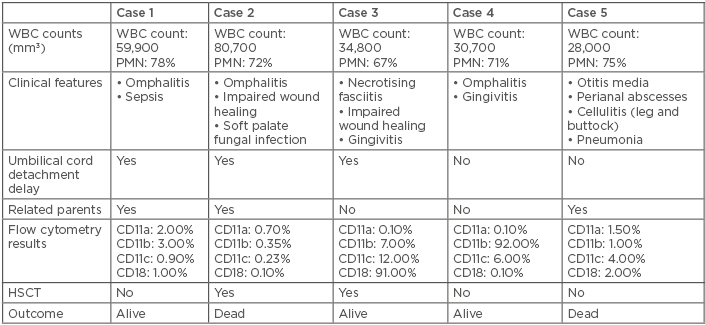
Table 1: Clinical features, laboratory findings, treatment, and outcome in five patients with leukocyte adhesion deficiency type-1.
HSCT: haematopoietic stem cell transplantation; PMN: polymorphonuclears; WBC: white blood cell.
HSCT was not available for all of the patients as a curative therapy. It has been performed in two patients, which led to death in one of them due to infection and sepsis because of the immunosuppressive state after receiving HSCT. The procedure was carried out in a tertiary centre for transplantation. Therefore, the authors do not have detailed information regarding post-transplant patient history.
DISCUSSION
LAD syndromes are rare genetically determined immunodeficiencies with challenging clinical manifestations.8 Here, the authors presented five cases with LAD with different presentations, management, and prognoses.
Jasani et al.23 reported a case of severe LAD-1 presented during the neonatal period and considered LAD-1 a rare neonatal presentation, with only two cases reported in the literature prior to 2014. Neonatal presentation of the disease is a function of disease severity. However, the age at presentation in the patients reported here was within the neonatal period in almost all of the cases (four at the age of 1 month and one at the age of 2 months).
Recurrent, severe, and difficult-to-treat bacterial infections4 are the predominant clinical manifestation of these patients, sometimes presenting as life-threatening infections, such as septicaemia, bronchopneumonia, and aseptic meningitis.24 Skin and mucosal surface infections may present as indolent and necrotic lesions that can enlarge and recur, commonly leading to systemic spread of infections.3 Infections usually appear first in the neonatal period as omphalitis and delayed umbilical detachment.5,25 Umbilical cord complications were more frequent in patients with severe LAD, as identified in a comprehensive review performed by Novoa et al.,11 and they reported a significant correlation between lack of these complications and survival to 2 years of age.
After infancy, gingivitis, periodontitis, and impaired wound healing4,26 are characteristic of LAD. Absence of pus formation is significant24 due to the impaired migration of leukocytes and defect in extravascular accumulation of polymorphonuclears and monocytes. Failure in clearing apoptotic polymorphonuclears and generating inflammatory modulator signals leads to impaired wound healing due to dysregulated macrophage–neutrophil interactions.27,28
Novoa et al.11 reported infections in 77% of their patients with severe LAD, of which the majority were due to respiratory infections, sepsis, and otitis media. Those with less severe deficiencies (CD18 ≥2%) in the previous study presented with periodontal infections, otitis media, and sepsis.11 Dababneh et al.29 reported periodontal findings, including intense redness and inflammation of the gingiva, primary teeth, and permanent dentition involvements, leading to rapid periodontal destruction refractory to conventional non-surgical therapies. However, the most frequent infections in our patients who had predominantly presented with severe LAD were omphalitis and delayed umbilical separation followed by oral ulcers indicating gingivitis. The authors could not find a significant relationship between the WBC counts and the severity of the disease (CD18 <2%) in the patients in this study.
The most frequently isolated micro-organisms from LAD patients are Staphylococcus aureus and Gram-negative bacteria.3 Other micro-organisms, such as fungal infections, although unusual, have also been reported.3,16,30 Lymphocyte extravasation is not affected; therefore, defences against viral infections are usually maintained.3 Despite severe infections in the patients in this study, the authors could not isolate micro-organisms from the patients because they had received long-term antibiotic therapies due to their severe infections every time before their admission. Fungal infection in the second case is assumed to be the result of phagocytic dysfunction. In this case, a positive fungal culture was not obtained. However, after an infectious consultation and because of the pattern of the lesions, the authors conducted an empirical therapy with an antifungal agent (amphotericin), which was accompanied by a good response to the therapy and the lesions disappeared.
Persistent neutrophilia in the absence of infections and dramatically increased myeloid leukocyte counts in the presence of infections are characteristic.4,31 Although a higher median WBC count were reported by Novoa et al.11 in 143 cases with CD18 <2% (48 versus 30×109/L), a limited correlation was found between CD18 expression and WBC counts for the entire cohort.
Patients with severe LAD-1 can present with a failure to thrive due to general malnourishment and sometimes colitis.3 In this study, the failure to thrive observed in children was because of malnourishment and recurrent infections. A rare mutation with non-functional although normal β2 integrins expression has been reported.32-34
Patients with moderate LAD mostly survive childhood.11 Severe LAD-1 is associated with significant mortality (reported as 75% by the age of 2 years)20,35 in those who do not receive allogeneic HSCT. Four of the patients reported, except one, fell into the classification of severe LAD-1. Two of these patients with severe LAD-1 are still alive while the other two died. HSCT was not available for all of the patients and one of the two patients who underwent HSCT died as a result of complications of the procedure. The children were not transplanted in the same centre where their disease was diagnosed; they were referred to a tertiary centre for bone marrow transplant. The cause of death in the transplanted child was fulminant sepsis. Unfortunately, due to the prolonged time taken searching for a matched donor, the patients experienced the complications of a severe chronic disease with recurrent infections, which significantly affected their prognoses even after receiving HSCT and was fatal for one of the patients.
Although ITGβ2 gene sequencing and mutation analysis may confirm the diagnosis and is useful for genetic counselling,35,36 none of the patients in this study had a mutation analysis and all of them were diagnosed based on their typical clinical manifestations and in vitro diagnostic tests, including CBC and blood flow cytometry.
CONCLUSION
The findings reported indicate that LAD-1 remains a life-threatening condition with omphalitis, oral, skin, and respiratory tract and ear infections being the most common complications. The most important issue in the management of these patients is to control infections. Early identification of these patients is essential in definitive diagnosis and early implementation of HSCT. However, HSCT was not associated with a favourable response in one of the two patients who received this treatment in the series.





