Meeting Summary
Improving therapy for people with allergies is a continuously evolving area and, due to the increasing prevalence of allergic diseases, options such as pharmacotherapy and allergen avoidance are inadequate alone to control these diseases. Worldwide, approximately 400 million people are affected by allergic rhinitis (AR) and 300 million people by asthma.1 Unlike anti-allergy medications, a unique feature of allergen immunotherapy (AIT) is that it modifies the underlying cause of disease,2 suggesting that it may be an optimal treatment approach. Guidelines, such as those from the European Medicines Agency (EMA) in 2009,3 provide the basis for optimising trial design for the development of new AIT preparations. A wide range of treatment modalities, including recombinant allergens, have been developed, and results from several studies, some only published in trial registries, provide clarity and insights into optimising clinical trial design even further.4-13 Lessons learned from these studies, which are scientifically informative for the community, were explored in this session.
In addition, the latest results were discussed from a dose-finding trial and a Phase III trial of a new allergoid treatment in development for patients with house dust mite (HDM)-induced asthma with or without AR or allergic rhinoconjunctivitis (ARC).14,15 Since AIT is recommended to be administered for 3 years, successful AIT requires adequate patient adherence over the long-term. The last section of this review focusses on strategies to optimise existing AIT and patient care, with a particular emphasis on reducing the number of injections during dose escalation when performing subcutaneous immunotherapy (SCIT) using pollen allergoids.
Development of Recombinant Allergen Immunotherapy: Lessons Learned
Professor Jörg Kleine-Tebbe
AIT, a well-known treatment option for immunoglobulin (Ig)E-mediated AR, ARC, and allergic asthma, is impeded by a lack of standardised extracts for many allergens and batch-to-batch variation in allergen concentration within available extracts because of the use of natural sources.16 Therapies that use proteins synthesised with recombinant DNA technology provide the opportunity to administer fully characterised molecules with reliable pharmaceutical quality in consistently identical vaccines.16-18 This unique approach has sparked much interest because it offers the potential to tailor vaccines to the specific major allergens of each patient, facilitates more precise administration of optimal allergen doses, and it allows the structure of IgE-binding allergen epitopes to be modified.16
All AIT products must undergo rigorous assessment before clinical use. The 2009 EMA guidelines for the development of AIT products to treat allergic diseases require early-phase studies to evaluate safety and tolerability, studies designed to establish a dose–response relationship for clinical efficacy, and at least one confirmatory trial using a randomised, double-blind, placebo-controlled design.3
In the early 2000s, a clinical trial programme of fully characterised, standardised, high-quality recombinant products for SCIT was initiated. The development programme for a recombinant grass allergen mix (rPhleum, Allergopharma GmbH & Co. KG, Reinbek, Germany) of equimolar concentrations of five different major allergens (Phl p 1, Phl p 2, Phl p 5a, Phl p 5b, and Phl p 6) from Phleum pratense (Timothy grass) consisted of five studies performed between 2003 and 2009 (Figure 1).4-9 Four studies performed between 2003 and 2010 (Figure 1) made up the development programme for the hypoallergenic recombinant folding variant (FV) of the major allergen Bet v1 of Betula verrucosa (rBet v1-FV).10-13 The results of these studies, some of which are so far only reported in clinical trial registers, can aid in understanding the nature of recombinant AIT and improving clinical trial design, enabling optimal selection of patients and endpoints. Most of the studies had a baseline period (1 year of observation to confirm a sufficient level of symptoms and evaluate medication use before randomisation) and a 2-year treatment period. This allows calculation of the change in efficacy between baseline and after 1 or 2 years of AIT, as well as the placebo effect. This contrasts with most AIT trials, which do not have a baseline period.
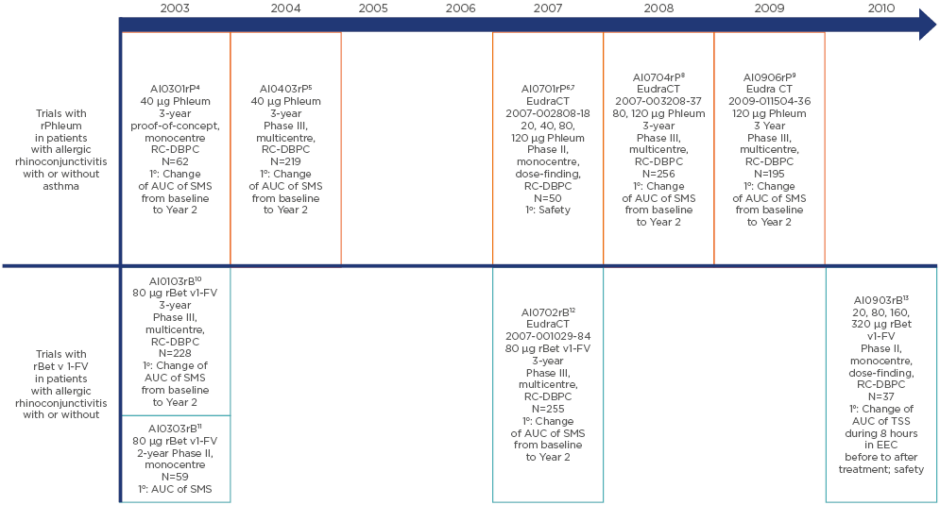
Figure 1: The clinical development programmes, including study design, patient numbers, doses, and primary endpoint, of the recombinant Phleum pratense (Timothy grass) pollen allergen mix (rPhleum) and the recombinant folding-variant of the birch (Betula verrucosa) pollen major allergen Bet v 1 (rBet v 1-FV) preparations.
1o: primary endpoint; AUC: area under the curve; DBPC: double-blind placebo-controlled; EEC: environmental exposure chamber; RC: randomised controlled; SMS: combined symptom medication score; TSS: total symptom score.
SCIT with rPhleum
The first proof-of-concept study (AI0301rP) for this preparation evaluated a 40 µg dose of rPhleum in 62 patients with grass pollen allergy.4 Symptom medication score (SMS) was 4.6 for active treatment versus 7.5 for placebo after 2 years (38.5% difference; p=0.051 [full analysis set]). Active treatment led to increases in IgG1 (60-fold) and IgG4 (400-fold) P. pratense- specific antibody concentrations, whereas specific IgE concentrations were significantly lower than with placebo. Overall, rPhleum demonstrated clinical efficacy in the per protocol set (p=0.044), was well tolerated, and induced strong allergen-specific IgG responses.
The Phase III AI0403rP trial,5 using the same dose (40 µg) of rPhleum, found improvement in SMS with both active treatment and placebo after 2 years, but the difference between the groups was not significant (data on file). The safety of increased doses was therefore investigated in the dose-finding study AI0701rP,6 which reported no systemic reactions with placebo, two with the 20 µg allergen mix, and three each for the 40, 80, and 120 µg allergen mix, indicating the safety of rPhleum even at high doses.7 The subsequent Phase III randomised trials AI0704rP,8 investigating 80 and 120 µg rPhleum, and AI0906rP,9 using 120 µg rPhleum, also found no significant difference between active treatment and placebo in change in SMS after 2 years (p=0.4153 and p=0.1124, respectively). In summary, rPhleum lacked clinically convincing data despite promising results early in the development programme. Being a fixed cocktail, it may not have been as effective in all patients due to individual sensitisation profile heterogeneity.
SCIT with rBet v1-FV
The preliminary AI0103rB trial investigating 80 µg of rBet v1-FV found a significant difference in change of SMS between active treatment and placebo after 2 years (p=0.014).10 The second trial (AI0303rB)11 compared the efficacy of 80 µg rBet v1-FV with a registered native birch pollen preparation. After 1 year of SCIT, SMS was lower for rBet v1-FV; however, the difference was not maintained at 2 years, with both treatments achieving reduced SMS scores. Therefore, rBet v1-FV may act more rapidly than SCIT with a native preparation, but ultimately the efficacy is equal. In 2007, the Phase III AI0702rB trial12 found no significant difference between 80 µg rBet v1-FV and placebo in change of SMS (p=0.1094). Lastly, a Phase II dose-finding study (AI0903rB)13 investigating doses of up to 320 µg rBet v1-FV found that, in an exposure chamber, total changes in symptom score from before to after SCIT for 10 weeks were significantly decreased and the level of IgG1 significantly increased in all active groups versus placebo. However, a clear dose–response relationship was lacking.13 The programme concluded that rBet v1-FV was more effective than placebo but not necessarily more effective than approved SCIT products derived from native birch pollen extracts.
Lessons Learned
Overall, recombinant preparations were neither more effective nor safer than already available preparations. However, negative results are not necessarily failures, and the data gathered is scientifically informative for the community, providing the opportunity to test new alternatives and improve trial design.
Firstly, these trials showed that allergic individuals display wide heterogeneity, which can obscure conclusions; raw data from these types of trials are useful to gauge the heterogeneity of the cohorts involved and should be made publicly available. Secondly, an inherent challenge of Phase III trials is to demonstrate specific effects that exceed nonspecific placebo effects. This can be hindered by regression to the mean; extreme observations tend to move towards the mean because subjective expectations influence results, and the natural course of the disease may result in changes in symptom burden during therapy and the Hawthorn effect (under observation, subjects behave differently). Thirdly, current endpoints required by EMA guidelines appear suboptimal; for example, SMS is limited by its subjective nature. To improve accuracy, SMS can be combined with other validated immunological parameters. Lastly, it is difficult to standardise exposure to therapies, the most accurate method for diagnosis remains uncertain, and there is a paucity in knowledge of what happens to recombinant peptides following administration.
These general problems have also occurred with other recombinant candidates, resulting in a halt in development. These include recombinant Bet v 1 sublingual immunotherapy (SLIT),19 a recombinant Amb a 1-CpG construct (ragweed pollen antigen),20 and two fairly long Fel d 1 peptides (which caused late-phase side effects).21 The recombinant grass pollen construct BM32, first tested in an exposure chamber, was well tolerated and achieved significant reductions in total nasal symptom score at 20 µg (p=0.03) and 40 µg (p=0.003).22 However, a recent Phase II field study found no significant difference between BM32 and placebo.23 Results from the Phase III trial for this product are anticipated for confirmation of efficacy.
With these new learnings in place, research is now focussing on AIT products that more accurately resemble natural allergens, which are available on the market and have confirmed efficacy and safety. Knowledge gained from this research may enable successful development of recombinant therapy principles in the future.
House Dust Mite SCIT: Focus on Patients with Asthma
Professor Marek Jutel
HDM sensitisation is important in AR, in which 49% of patients are HDM-sensitised, and in allergic asthma, with 50–85% of patients being HDM-sensitised.24-27
Current Guidelines for Allergen Immunotherapy in Allergic Asthma
Thus far, there is little guidance available on AIT in allergic asthma. However, an expert working group of the European Academy of Allergy and Clinical Immunology (EAACI) are currently preparing more complete recommendations using the AGREE II international tool.28 The group recommend SCIT or SLIT for adequately controlled mild-to-moderate disease and AIT should be implemented to reduce symptoms, improve quality of life, and minimise future risk.28 SCIT has been found to significantly reduce symptoms and medication use but for HDM SLIT there is only weak evidence available for achieving asthma control and moderate evidence for decreasing exacerbations.28
Unmet Needs in House Dust Mite Allergen Immunotherapy
Unmet needs in HDM AIT include a lack of adequately powered, randomised, controlled studies and well-characterised allergen preparations. Management of exposure monitoring is a major limitation of HDM studies due to uncertainty around patients collecting samples correctly and how to account for differences between households. Allergen exposure chambers may be more accurate but require validation in Phase III studies. Managing exposure is easier for pollen studies; pollen chambers, which can be very useful in paediatric studies, should be used to gain an understanding of immunotherapy efficacy and the identification of biomarkers that need to be validated. Another large unmet need is well- defined outcome measures in HDM-induced asthma; a potential outcome measure is the reduction of inhaled corticosteroid (ICS) use while retaining asthma control. In a total of 65 HDM-allergic children and adolescents (aged 6–17 years) with controlled bronchial asthma (mild-to-moderate, as classified by the Global Initiative for Asthma [GINA])29 requiring ICS, after 1 year of HDM SCIT, mean fluticasone propionate dose decreased to 190 µg/day, below the level thought to result in growth suppression (200 µg/day). At baseline, all patients required ICS but after the first, second, and third years, 30.3%, 54.5%, and 60.6% of patients, respectively, no longer required ICS.30
Dermatophagoides pteronyssinus Allergoid
For SCIT, an aluminium hydroxide-adsorbed depot allergoid preparation of standardised, high concentration Dermatophagoides pteronyssinus allergens modified with formaldehyde and glutaraldehyde has been developed.31 AL1009ac, a dose-finding study of this HDM allergoid, included adult patients (aged 18–40 years) with HDM-induced asthma with or without AR/ARC and requiring ICS (fluticasone equivalent; maximum daily dose of ≤500 µg).14 The primary endpoint was late-phase response 6 hours after intracutaneous testing (IC) with D. pteronyssinus extract. Secondary endpoints included early-phase response 20 minutes after intracutaneous testing, minimal asthma control dose of ICS, and safety. Overall, 146 patients were randomised to placebo or to 600, 1,800, 3,000, or 5,400 protein nitrogen units (PNU)/mL of HDM allergoid.
A significantly reduced late-phase IC response was observed with all doses compared with placebo (p<0.001). In patients with mild-to- severe asthma, as classified by GINA,29 a significant reduction in swelling area was reported in all dose groups versus placebo.14 Compared with placebo, statistical significance in the reduction of early-phase IC response was only achieved with 3,000 PNU (p<0.01) and a significant difference in the number of patients who did not need ICS after treatment was only found with 5,400 PNU (6 versus 11 patients; p<0.05; unpublished data). From baseline to post treatment in the 5,400 PNU group, no patients increased their ICS dose and 69% of patients had a dose reduction by two steps (unpublished data). The proportions of patients with at least one adverse event (AE) related to study medication were 6.3%, 16.7%, 19.4%, 14.3%, and 35.5% in the placebo and 600, 1,800, 3,000, and 5,400 PNU groups, respectively.14
Overall, the most effective doses were 3,000 and 5,400 PNU, with 5,400 PNU being more effective in the reduction of ICS needed to retain asthma control. All tested doses were well tolerated, although more AE were observed in the 5,400 PNU group, but with no greater severity.14 Therefore, 5,400 PNU was chosen as the dose with the most favourable benefit–risk ratio for further investigation.
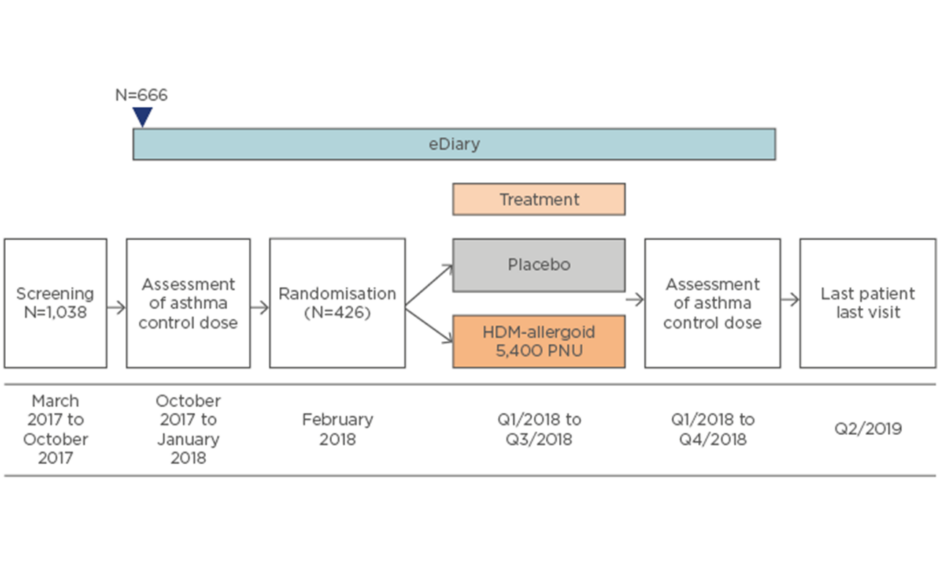
AL1402ac15 is an ongoing Phase III, multicentre, randomised, double-blind, placebo-controlled clinical trial evaluating the efficacy and safety of SCIT with this investigational allergoid in patients with allergic bronchial asthma with or without AR or ARC (Figure 2). Currently, 1,038 patients have been screened, 666 have entered details into their eDiary at baseline, and 426 have been randomised. The primary endpoint is the change between baseline and completion of AIT in dose steps of the minimum daily ICS (budesonide) dose required to ensure asthma control according to an asthma control questionnaire (ACQ6; score ≤1.0). Secondary endpoints include quality of life, combined rhinitis SMS, and the first timepoint at which moderate or severe asthma exacerbation is noted.
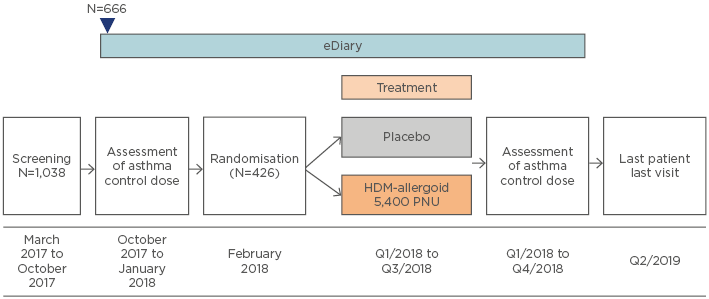
Figure 2: Study design of the AL1402ac Phase III clinical trial evaluating safety and efficacy of subcutaneous immunotherapy with the house dust mite allergoid in patients with house dust mite-allergic asthma and allergic rhinitis or rhinoconjunctivitis.15
HDM: house dust mite; PNU: protein nitrogen units/mL; Q: quarter.
Overall, investigational HDM allergoid SCIT is effective and well tolerated, and the dose with the most favourable benefit–risk ratio has been identified. Outcome measures of asthma AIT trials should be harmonised, with ICS reduction or asthma exacerbations being preferable parameters.
Shortcuts in Pollen SCIT: A Step Towards Better Patient Care
Professor Matthias Kopp
When considering the future of AIT and improving patient care, there are numerous potential strategies, including development of new SLIT products, application route optimisation, use of purified allergens, and development for other indications (e.g., food allergy). Two particularly promising avenues are initiation of pollen AIT during the respective pollen season and accelerated dose escalation.
Successful AIT is impeded by a lack of patient adherence.1,32 AIT is highly effective in AR and allergic asthma and is known to modify the underlying cause of the disease.2,32 AIT has been shown to have a long-lasting benefit, can prevent the onset of new sensitisations, and can prevent asthma onset in children with AR/ARC.32 Patient adherence can be improved by good communication with the patient, educational programmes for patients and healthcare providers, and improving AIT convenience.1,32
Accelerated dose escalation may reduce the requirement for doctor visits, thus increasing patient adherence.32 Moreover, accelerated dose escalation is possible without increased number and/or severity of AE, and AIT can be initiated throughout the year, even during the pollen season. Additional data have recently become available for SCIT with birch and grass pollen allergoids.
Birch Pollen Allergoid
The Phase II, open-label ASTOR trial33 compared the safety and tolerability of accelerated (four injections, n=63) and standard (seven injections, n=67) dose escalation schedules of birch pollen allergoid in adult patients (18–65 years) with seasonal AR with or without controlled asthma. The primary outcome was incidence of systemic AE related to SCIT, graded according to the World Allergy Organization (WAO) system.34 Overall, 57.1% of accelerated and 58.2% of standard scheme patients experienced at least one AE, with local AE being experienced by 54.0% and 56.7%, respectively. At least one Grade I–II systemic AE was reported in 6.3% of accelerated and 3.0% of standard scheme patients. No Grade III–IV systemic or serious AE were observed. Overall, 85.5% of accelerated and 95.4% of standard group patients rated the tolerability of their treatment as ‘good to very good’.35
Grass Pollen Allergoid
The Phase III randomised, open-label SuBITo trial36 compared the safety and tolerability of intraseasonal (n=158) and standard preseasonal (n=73) dose escalation schedules of the six-grasses allergoid. At least one AE was experienced by 68.4% of intraseasonal and 56.1% of preseasonal patients. Incidence of local AE was not statistically different between intraseasonal (64.6%) and preseasonal (54.8%) patients (p=0.1907), with most events being mild (intraseasonal: 55.9% versus preseasonal: 60.0%) or moderate (intraseasonal: 36.3% versus preseasonal: 30.0%). At least one systemic Grade I–II AE was observed in 3.2% of intraseasonal and preseasonal patients. Tolerability was rated as ‘good to very good’ in 85.0% of intraseasonal and 88.6% of preseasonal patients.37 These results indicate that starting SCIT during the season is appropriate, with timing being less important than whether or not SCIT is started, since clinical benefit does not occur until the year after initiation and delays to initiation may result in the patient electing not to begin AIT at all.
A previous Phase II trial38 assessed accelerated dose escalation of a six-grasses pollen allergoid preparation consisting of four weekly injections (200, 600, 2,000, and 6,000 TU/mL). No difference between accelerated and standard escalation regarding the intensity and number of local and systemic AE was found. This raises the question of whether further increases in the rate of escalation would provide additional benefits. It is hypothesised that further escalation from the four weekly injections to three injections (1,000, 3,000, and 6,000 TU/mL) using only the 10,000 TU/mL vial will allow patients to reach the cumulative dose earlier and minimise risk of confusion between different strength vials.
This is being investigated in the ongoing Phase II ONSeT trial,39 underway at 13 European sites. The study aims to investigate the safety and tolerability of standard (seven injections) and ‘high-speed’ (three injections) escalation schedules of the six-grasses allergoid in adult patients (aged 18–65 years) with moderate-to-severe seasonal AR or ARC with or without asthma (Figure 3). Preliminary results were presented during the EAACI 2018 Congress.40
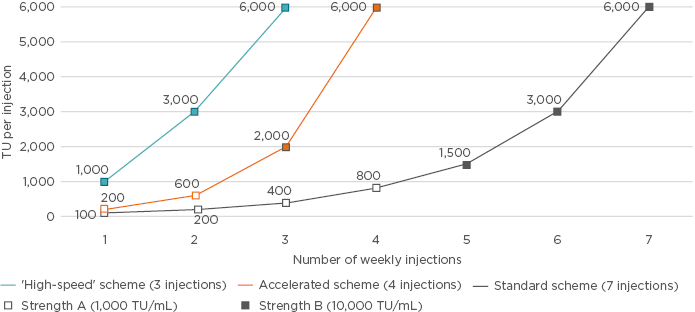
Figure 3: Different dose escalation schemes tested in clinical trials using pollen allergoids with the standard scheme (grey), consisting of seven injections of strength A (1,000 TU/mL) plus B (10,000 TU/mL);33,35,38-40 the accelerated scheme (orange), consisting of four injections of strength A plus B;33,35,38 or ‘high-speed’ scheme (blue), consisting of three injections of strength B.39,40
TU: therapeutic units.
Findings and Future Investigation
Accelerated dose escalation of grass and birch pollen allergoids demonstrated comparable safety and tolerability in adult patients with AR with or without asthma. However, patient numbers in individual trials were low. When combining the ASTOR,33,35 Chaker et al.,38 and ONSeT39,40 preliminary data (N=338), there was no significant difference in local AE after accelerated, ‘high-speed’, or standard dose escalation, but a higher number of Grade I–II systemic AE in the accelerated and ‘high-speed’ schemes was found.33,35,38-40 No Grade III–IV systemic AE were observed with any of the escalation schedules.
Conclusion
These clinical development programmes emphasise that a significant challenge of Phase III trials is demonstrating specific effects beyond the universal placebo effect. Also, dose- finding Phase II trials are challenging since clear dose responses to all investigated endpoints are often not demonstrated. However, even negative results (i.e., no statistically significant superiority for active treatment versus placebo) are scientifically informative, allowing improvements in future trials. Investigational HDM allergoid SCIT has shown efficacy and tolerability in adult patients with HDM-induced asthma with or without AR/ARC in a dose-finding trial, and the 5,400 PNU dose has been selected for further investigation. Large studies, including the ongoing Phase III AL1402ac trial,15 should greatly improve our understanding of AIT in asthma. Accelerated dose escalation with grass and birch pollen allergoids has displayed comparable safety and tolerability to standard escalation schedules in adult patients with AR with or without asthma, resulting in reductions from 6 to 2 weeks of escalation. Initiation of SCIT with a standard regimen of the grass pollen allergoid is well tolerated in adults during the grass pollen season. Accelerated dose escalation is expected to attract more patients to SCIT and to increase adherence.





