
Meeting Summary
Hereditary angioedema (HAE) is a rare autosomal dominant condition caused by a deficiency or dysfunction of C1 esterase inhibitor (C1-INH) that normally blocks activation of C1, the first component of the complement cascade. The condition manifests as recurrent self-limiting episodes of angioedema, without urticaria or pruritus, most commonly affecting the skin or mucosal tissues of the upper respiratory and gastrointestinal tracts.1 Symptoms are disabling and can be life-threatening when affecting the upper airways.1 Low awareness of the condition and its resemblance to other disorders typically leads to delays in diagnosis.2 Multiple mutations of the human C1-INH gene (SERPING1) have been identified, some of which cause HAE and some of which do not.1 Genetic testing alone is therefore not diagnostic of HAE and needs to be supplemented with biochemical testing and hereditary information. There are mixed opinions among clinicians and scientists on the utility of genetic testing for diagnosis of HAE. The objective of this symposium was to raise awareness of HAE and its diagnosis, along with the role of genetic testing, familial testing, and future diagnostic methods for this disorder.
Dr Stephen Jolles chaired the symposium and opened with a presentation on current diagnosis of HAE. Prof Marco Cicardi presented on biomarkers that enable earlier diagnosis of HAE; and in the final presentation, Dr Coen Maas discussed the future of HAE diagnosis. Interactive voting and question and answer sessions were used to elicit the opinions of the audience at intervals throughout the symposium, which was concluded with a general discussion session.
Diagnosing Hereditary Angioedema Patients Today
Doctor Stephen Jolles
Initial diagnosis of HAE centres around clinical presentation and physical findings. Patients typically present with self-limiting episodes of angioedema (without urticaria or pruritus), which most commonly affects the skin or mucosal tissues of the upper respiratory and gastrointestinal tracts. Diagnosis is supported by biochemical testing that should ideally be conducted twice, with at least 1 month between tests. A family history of HAE is supportive of diagnosis, but not required. HAE affects approximately 1 in 50,000 people, with equal prevalence in men and women.3-5 Unfortunately, diagnosis is often delayed, typically by 10–18 years.3-5 Type I HAE is the most prevalent, accounting for about 85% of cases, and is characterised by low C1-INH and C4 levels.3-5 Type II HAE (15% of cases) presents with normal or elevated but dysfunctional C1-INH and low C4 level.3-5
HAE has an autosomal dominant inheritance pattern, with one copy of the affected gene being sufficient to cause the disorder. HAE can also arise from spontaneous mutations in people with no family history of the disorder, accounting for 20–25% of HAE cases.6 Inherited forms of HAE are caused by a mutation in the SERPING1 (serine proteinase inhibitor clade G) gene, which is mapped to chromosome 11 (11q12–q13.1). SERPING1 contains eight exons and seven introns and is prone to large deletions and duplications.7 Type I HAE is caused by mutations throughout SERPING1 that results in impaired protein folding or secretion.7 Mutations in Type II HAE involve the active site of exon 8, which results in a dysfunctional protein that impairs binding to proteases.7 Interestingly, about 10% of people with SERPING1 mutations are asymptomatic.7
Over the years, a variety of different methods have been used for the diagnosis and classification of HAE, including whether or not a particular drug is successful in treating idiopathic histaminergic angioedema; biochemistry and immunology testing of acquired C1-INH deficiency; biochemistry exclusion (+/-) of angiotensin-converting enzyme (ACE)-induced acquired angioedema (AAE); history and biochemistry of Type I and Type II HAE; genetics of hereditary Factor XII (FXII) angioedema; and biochemistry and genetics of normal C1-INH-HAE. Different tests are therefore required to reach a diagnosis depending on the setting. In routine immunology and biochemistry laboratories, protein (C3, C4, C1-INH) and functional (fC1-INH) tests are generally available. Protein tests require a serum sample, measured by nephelometry or radial immunodiffusion, which, although may be subject to interference with lipidaemia, haemolysis, or icterus, are generally very reliable.8 Functional tests (required for Type II HAE diagnosis) are performed on serum samples by sandwich enzyme-linked immunosorbent assay (ELISA) (of activated C1s); the main issue with this test is sample delay, so fresh samples need prompt freezing if transport is needed and a delay before analysis is anticipated.8 New functional tests in development aim to mirror more closely pathways of interest in HAE and use biotinylated activated FXII (FXII-a) or biotinylated kallikrein bound to avidin-coated plates for the detection of bound C1-INH.8
There are several additional causes of low C4 other than HAE. These include AAE, systemic lupus erythematosus, vasculitis, antiphospholipid syndrome, cryoglobulinaemia (essential or secondary, e.g. to hepatitis B virus and hepatitis C virus), cold agglutinin disease (Immunoglobulin M red blood cell), C4 nephritic factor (C4b2a), viral infections with parvovirus, genetic null alleles (C4a and C4b), and rarely, advanced liver disease.9 In AAE, the expected patient profile would be low C4, low C1q, both antigenic and functionally low C1-INH, and anti-C1 INH antibodies (in 70% of patients). AAE is a disease with late onset (>40 years of age) and there is usually no family history of the disorder.10,11
There are two categories of HAE with normal C1-INH-HAE with FXII mutations and without FXII mutations.4 HAE with normal C1-INH presents predominantly in adulthood and may have more frequent involvement of the tongue, uvula, and face, but there is no erythema multiforme.4 Idiopathic angioedema needs to be excluded as this disorder also presents with normal C1-INH.4
In conclusion, clinical history, physical examination, and supportive family history are central in the diagnosis of HAE. Frontline biochemical testing involves C4, C1-INH, and fC1-INH, optimally repeated a month or more apart. In patients with normal results, there may be an additional role for testing during an attack, because a normal C4 during an attack in HAE is an extremely rare event. Some current diagnostic tests may have utility in therapeutic monitoring, but are unable to predict disease severity or explain variability between patients.
During his presentation, Dr Jolles asked the audience whether or not genetic testing was available to them in their practice. A total of 56% of the audience responded ‘yes’, while 44% do not have genetic testing available. Dr Jolles then asked the audience the most common reason for them conducting genetic testing: the majority (50%) voted for diagnostic uncertainty; 14% for routine part of work-up; 14% for family testing; 6% to rule out HAE; and 16% never use genetic testing.
Biomarkers: Improving Diagnosis
Professor Marco Cicardi
There are various approaches available to diagnose angioedema. These include the identification of aetiology, pathogenic mechanisms, and phenotypes, and the stratification of phenotypes.
Identification of Aetiology
Angioedema due to C1-INH deficiency was identified by Virginia Donaldson as the biomarker that defines a group of patients with the phenotype of recurrent angioedema. Two types of angioedema due to C1-INH deficiency exist: the inherited autosomal dominant form (HAE), and the non-inherited form caused by anti-C1 INH antibodies (AAE) and/or lymphoproliferative disease.12 HAE due to a mutation in the FXII gene (known as the Thr328Lys mutation) has been traced back to a gene rearrangement in a single person. People with this type of HAE have a different phenotype to those with HAE due to C1-INH deficiency.13,14 Patients with angioedema due to deficiencies in ACE can be identified by the pattern of relapses after discontinuation of ACE-inhibitor therapy.15
Identification of Pathogenetic Mechanisms
Bradykinin is the main mediator of angioedema.16 In HAE with FXII mutations, FXII mutant proteins are cleaved and rapidly activated by plasmin and escape inhibition by C1-INH, thereby causing excessive bradykinin formation.17 In ACE-inhibitor-related angioedema, catabolism of bradykinin is reduced due to inhibition of ACE (which degrades bradykinin). Overall levels of bradykinin are increased, but cleavage of the precursor to bradykinin (high molecular weight kininogen) is not increased.18,19 Idiopathic histaminergic angioedema is diagnosed when angioedema without wheals stops recurring upon continuous treatment with an H1 histamine receptor blocker (up to 4-times the approved dose).3
Identification of Phenotypes
Phenotypes can be used to diagnose or sub-categorise groups of patients. In a nationwide survey of 983 Italian patients with HAE (87% with Type I and 13% with Type II), a diagnosis of C1-INH deficiency could be made based on reduction in C1-INH function (median value: 20% for Type I and 19% for Type II).20 Antigenic C1-INH, however, was within the normal range for Type II patients (median value: 96%) but not for Type I patients (median value: 21%), demonstrating the value of this technique in categorising Type I and Type II patients.20 In patients with FXII mutations, there is a large difference in phenotype, as male patients rarely experience oedema whereas female patients almost always do.21
Several different forms of angioedema can therefore be identified. Diagnosis of acquired idiopathic non-histaminergic angioedema is based on the exclusion of C1-INH deficiency, ACE inhibitor therapy, evidence for causative agents, absence of family history of angioedema, and absence of mutations in the FXII gene.22 In addition, there are two groups of patients, those that respond to anti-histamine treatment and those who do not.23 Bradykinin has been shown to be an essential mediator in most subtypes of angioedema without wheals (Figure 1).22
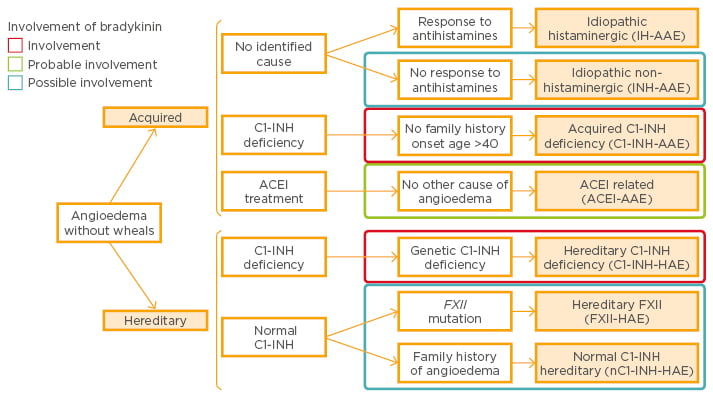
Figure 1: Classification of angioedema without wheals and involvement of bradykinin.22
ACEI: angiotensin-converting enzyme inhibitor; FXII: Factor XII; INH: esterase inhibitor; AAE: acquired angioedema; HAE: hereditary angioedema.
Stratification of Phenotypes
Recurrent angioedema without urticaria is associated with degradation of bradykinin, which is broken down (cleaved) by kallikrein.12 Cleavage results in two-chain high molecular weight kininogen molecules that can be visualised by electrophoresis and used as a biomarker for different forms of angioedema.23 In patients with C1-INH HAE, kininogen is a potential biomarker for disease severity, as patients with fewer symptoms have lower levels of cleaved kininogen than patients who are more symptomatic.24 Several other proteins have been shown to be involved in the bradykinin-forming cascade, including the plasminogen activator urokinase and vascular endothelial-like growth factor (VEGF); VEGF could potentially be used as a biomarker to distinguish different forms of angioedema or disease severity.25-27
In conclusion, C1-INH deficiency, mutations in FXII, and treatment with ACE-inhibitors are diagnostic of specific forms of angioedema. Evidence of histamine as the mediator distinguishes one form of AAE. Other forms of angioedema are defined only by the clinical profile and the exclusion of specific diagnoses. Markers to stratify patients with angioedema are still only partially identified and need to be further refined.
Question and Answer Session
Prof Cicardi was asked if he could provide further insight into the classification of non-histaminergic angioedema, and responded that the important issue is that in these patients, a mechanism exists that is different from the mechanism in patients that respond to antihistamine. Histamine may play a role in these patients, but is not the main mediator.
When asked if the kinetics of C1-INH and fC1-INH differ upon administration of C1-INH in people with hereditary versus those with AAE, Prof Cicardi responded that the kinetics are very different within patients. Patients with C1-INH deficiency demonstrate much more rapid catabolism of C1-INH than healthy people.
In response to the question of whether or not high molecular weight kininogen can serve as a prognostic biomarker, Prof Cicardi responded that kininogen may serve as an acceptable biomarker in the future, but the technical application and sensitivity of the assay needs improvement.
Looking Towards a Future of Improved Risk Stratification and Management
Doctor Coen Maas
As presented by Prof Cicardi, bradykinin is an important disease mediator in HAE. Accurate detection of bradykinin production during an angioedema attack, however, is challenging as its production is localised and difficult to detect systemically.28 Bradykinin is also rapidly broken down by kinases, which makes detection even more difficult.
Bradykinin is the end-product of the contact activation system that results from cleavage from its precursor protein, kininogen, by kallikrein. This process results in the production of high molecular weight cleaved kininogen (Figure 2). High molecular weight cleaved kininogen may be a suitable biomarker for contact activation as it is insensitive to kinases and readily detectable in the systemic circulation of healthy subjects.
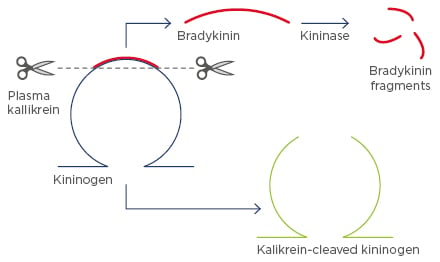
Figure 2: Cleavage of kininogen, producing high molecular weight cleaved kininogen.
In addition to cleavage by kallikrein, during an angioedema attack, kininogen is cleaved by several other enzymes and this may occur without the accompanied release of bradykinin. One of these enzymes is plasmin, and cleavage of kininogen by either kallikrein or plasmin results in similarly sized products when assayed by Western blotting.29 Evaluation of kininogen cleavage using this method is therefore not adequate to accurately measure bradykinin production, and an assay with small (17 kDa) highly-specific monoclonal antibodies known as nanobodies is being developed to accurately differentiate kallikrein- cleaved kininogen.30
FXII is the initiator of the contact activation system, and collective evidence points towards a role for FXII in angioedema attacks. This enzyme normally has several cleavage sites. People with mutations in FXII have an additional cleavage site in FXII, which causes the enzyme to fragment and activate more rapidly.17 In three types of FXII-HAE, mutants of FXII that are associated with angioedema were found to be particularly sensitive to activation by plasmin, resulting in higher levels of FXII. When no pathological mutations are present, several other enzymes may also be involved in FXII cleavage, including neutrophil elastase.30 Indeed, studies indicate that neutrophil elastase and kallikrein act synergistically to activate FXII.30
To summarise, kallikrein-cleaved kininogen is a circuit marker for bradykinin production and a potential surrogate marker for HAE. It is important, however, to understand that cleavage may not always represent bradykinin formation and the bradykinin-forming mechanism is best analysed under controlled conditions to prevent peri-analytical artefacts, tailored sampling procedures may be required. The study of FXII is key to understanding disease mechanisms in HAE, and may lead to identification of the underlying cause of attacks and, as yet, unidentified mechanisms in this disease.
Question and Answer Session
Dr Maas was asked if there was a marker for risk of angioedema attack and responded that studies investigating the natural triggers of FXII activation have demonstrated that plasminogen levels correspond well with disease activity.17 In patients with HAE and FXII mutations, the risk corresponds with the enzyme that triggers FXII. Different triggers may activate FXII in different patients. In general, however, bradykinin production is a good surrogate marker for disease activity.
A member of the audience asked Dr Maas if patients have co-existent histaminergic and bradykinin-activated HAE. Dr Maas responded that there is a mechanistically strong overlap between histaminergic reactions and the formation of bradykinin, and that this has been observed in patients with anaphylactic shock (without angioedema).31 In animal models, at least 50% of the hypotension in anaphylactic shock is directly attributable to bradykinin.32 Bradykinin may therefore be secondary to a histaminergic reaction in patients.
When asked if taking samples during an attack would improve the quality of analysis, Dr Maas responded that it would. However, where plasma samples are useful, serum samples have the caveat that they will be fully clotted and all kininogen will be cleaved. However, samples taken during an attack can be informative in detailing the difference between the active and inactive phases of angioedema.
Finally, to a question of whether biomarkers will be used to aid treatment of angioedema, Dr Maas responded that it was possible, but the amount of bradykinin required in patients with idiopathic angioedema needs to be elucidated first.
Discussion Q&A and Interactive Voting
Interactive Voting
Dr Jolles opened the session and asked the audience what proportion of family members have been tested in their clinical cohorts. There was wide variation in the proportion of clinicians that conduct genetic testing and those that do not. Only 17% of the audience have conducted genetic testing on 75–100% of family members, while 11% have conducted no genetic testing on family members.
Dr Jolles then asked the audience what proportion of their patients should have a genetic diagnosis of HAE. Just over 44% of the audience responded that >75% of their patients should have a genetic diagnosis, compared with 21% that thought that <10% should be tested.
Finally, Dr Jolles asked the audience what the main barriers to genetic testing were. The main barriers were cost (39% of the audience) and availability (34%). Difficulties in testing family members was voted by 18% of the audience, and only 9% thought it was unnecessary.
Discussion Q & A
Several questions were posed by members of the audience:
Q: How useful is genetic testing in patients with an HAE clinical phenotype and clinical manifestations, but who have normal C4, C1-INH, and fC1-INH?
Prof Cicardi responded that the diagnosis of C1-INH deficiency is based on a biochemical diagnosis, so genetic testing will not necessarily help these patients. It can, however, determine if the cause of C1-INH deficiency is genetic, which may be useful if there is no family history. A test for FXII could, however, be useful and may tell you why a patient has angioedema.
Q: Is biochemical testing in C1-INH deficiency sufficient for a diagnosis of HAE?
Prof Cicardi responded that for many patients, genetic testing is not useful. A particular level of C1-INH deficiency has to be reached before angioedema occurs and this cannot be predicted by genetic testing. Even if a mutation in C1-INH is demonstrated, it does not mean that the activity of the gene is modified.
Dr Jolles said that genetic testing could be useful for exclusion purposes in cases that are difficult to diagnose. In addition, novel mutations that are found and associated with an appropriate clinical presentation and biochemical results and that are then included in genetic databases would increase the probability of being able to more accurately diagnose HAE in the future.
Q: What should be the approach in patients in which a new mutation variation is found? And, secondly, in patients in which no mutations are found for C4 and C1-INH?
Prof Cicardi responded that segregation studies need to be conducted first to evaluate the relationship between any new mutation and the function of C1-INH, and, in the second case, Dr Jolles highlighted that genetic testing does have limitations in that only exons are generally evaluated; mutations in introns or upstream of the gene will not be detected.
Q: Should genotyping be the preferred diagnostic test for newborns and children?
Prof Cicardi responded that infants (<1 year of age) may have C1-INH levels that are inconclusive. In these patients, if there is already a family history of the disease, genetic testing may be the most precise method of diagnosis. After 1 year of age, C1-INH function can be used.
Symposium Summary
Dr Jolles summarised the presentations and discussion from the symposium:
- HAE is caused by C1-INH deficiency or dysfunction
- Current diagnosis is based on clinical presentation, C4 and C1-INH testing, and/or family testing: it is important that family members are encouraged to be tested for HAE
- Genetic testing is not routinely conducted, possibly due to lack of availability, cost, and that a firm diagnosis can be established in most cases of Type 1 and 2 HAE with biochemical testing
- Biomarkers could potentially help to identify triggers for HAE attacks and guide clinical treatment
- Kallikrein-cleaved kininogen is a potentially attractive surrogate marker for bradykinin formation, and therefore HAE
- Research developments are improving our understanding of the processes involved in HAE





