G protein-coupled receptors are tractable targets for pharmacotherapies;1 however, challenges arise when it is necessary to either deorphanise a known receptor by finding its ligand or identify the receptors responsible for mediating biological effects of particular ligands.2 One of these orphan ligands is Cxcl14, a chemokine that has been linked with obesity and glucose intolerance,3 although the molecular pathways underlying this association are poorly understood. Therefore, we investigated the role of Cxcl14 in islets by measuring its expression and quantifying its effects on beta cell function, and aimed to identify the signalling responsible for mediating these effects.
We identified that Cxcl14 mRNA was expressed in mouse islets, confirming our previous observation,4 and demonstrated that the expression levels were approximately 20-fold lower than those in brown adipose tissue. Fluorescence immunohistochemistry analysis of a mouse pancreas demonstrated that Cxcl14 was absent from islet beta cells but was present in the majority of delta cells. To study the functional profile of this orphan ligand, insulin secretion from MIN6 beta cells and mouse islets was quantified in the presence of Cxcl14, which demonstrated that Cxcl14 induced a significant concentration-dependent decrease in glucose-stimulated insulin secretion. These results are in agreement with the expression of Cxcl14 by delta cells, suggesting that it may be co-released with somatostatin to have paracrine effects and inhibit beta cell secretory function. During discussion of this presentation, it was suggested that further exploration could focus on whether there are any changes in Cxcl14 expression by delta cells of islets isolated from organ donors with Type 2 diabetes mellitus (T2DM). This is part of our ongoing research and it is possible that Cxcl14 overexpression could contribute to insulin secretory dysfunction in T2DM.
Cxcl14 may signal via a chemokine receptor, similar to other chemokines, so we quantified mRNA expression profiles of all chemokine receptors in MIN6 beta cells and mouse islets by quantitative PCR; this indicated that Cxcr4 and Cxcr7 are the most likely candidates for transducing the effects of Cxcl14 in beta cells. However, beta-arrestin recruitment experiments indicated that Cxcl14 did not activate either of these receptors, ruling out their involvement in mediating its signalling.
Analysis of downstream signalling by Cxcl14 indicated that it had no effect on cAMP generation in beta cells. However, experiments using 2-deoxyglucose, which is phosphorylated by beta cell glucokinase to non-metabolisable 2-deoxyglucose-6-phosphate, indicated that Cxcl14 caused a concentration-dependent inhibition of 2-deoxyglucose-6-phosphate generation in MIN6 beta cells and mouse islets. This suggests that Cxcl14 can inhibit glucokinase activity, and the expected reduction in ATP production by Cxcl14 was confirmed in experiments using mouse islets. It was suggested during discussion of these results that identification of beta cell mitochondrial activity in response to Cxcl14 would provide additional understanding of how this chemokine disrupts glucose metabolism.
In summary, our data reveal that Cxcl14 exhibits direct inhibitory effects on islet beta cells to reduce glucose-induced insulin secretion, and this is most likely a consequence of impaired glucose metabolism (Table 1). Thus, these beta cell-directed effects of Cxcl14 will contribute to the glucose dysregulation that occurs as a consequence of its upregulation in obese individuals and its induction of insulin resistance.3 These data highlight the utility of Cxcl14 inhibition as a possible therapeutic approach for T2DM.
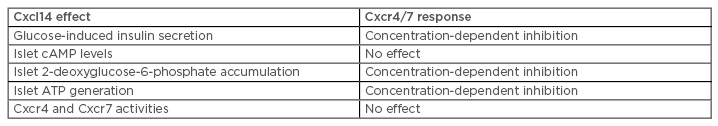
Table 1: Summary of the effects of Cxcl14 in islets and on Cxcr4/7 activities.