Abstract
Diabetic ketoacidosis (DKA) is an obstetrical emergency that is associated with an elevated risk of adverse pregnancy outcomes. This includes pregnancy loss in up to 1 in 3 cases. Due to the normal metabolic changes that occur during pregnancy, females who are pregnant are more vulnerable to DKA, and it can occur at lesser than expected degrees of hyperglycaemia. Presenting symptoms can be non-specific and include nausea and vomiting, fatigue, polydipsia, and polyuria. DKA may be the first presentation of previously undiagnosed diabetes. Therefore, high index of suspicion, along with prompt diagnosis and management, is essential. The cornerstones of management include intravenous insulin, intravenous fluids, and electrolyte replacement. Treatment generally follows the principles for DKA management outside of pregnancy, with some additional considerations. Close maternal and fetal monitoring is essential, and intensive care unit admission is typically required to adequately achieve this goal. In all situations, a thorough investigation should occur to address the underlying cause of the DKA and prevent further episodes. This review article outlines the potential etiopathogenesis, clinical presentation, and management of DKA in pregnancy.
Key Points
1. DKA in pregnancy is an obstetric emergency that leads to increased risk of maternal and fetal mortality without prompt diagnosis and management. Pregnant individuals with diabetes are at higher risk of developing DKA than non-pregnant individuals with diabetes, and DKA can occur at lower levels of hyperglycaemia.2.Timely recognition and management of DKA in pregnancy are crucial for optimising outcomes. Patients should be managed with intravenous insulin, intravenous fluids, and electrolyte replacement, and should have close maternal and fetal monitoring throughout.
3. Identifying DKA precipitants is important not only for treatment, but for reducing the risk of future recurrence. Improved understanding and evaluation could help with development of pregnancy-related DKA prevention strategies in the future.
INTRODUCTION
Diabetic ketoacidosis (DKA), characterised by the triad of acute hyperglycaemia, metabolic acidosis, and ketosis, is an acute life-threatening but preventable complication of diabetes.1,2 The Centers for Disease Control and Prevention (CDC) reported that the age-adjusted rate of DKA hospitalisation has increased by 54.9%, from 19.5 to 30.2 per 1,000 people, at an average annual rate of 6.3% from 2009 to 2014. The rates were highest in individuals less than 45 years of age. However, the in-hospital case-fatality rates have declined overall by 63.6%.1 Although DKA is typically encountered in patients with Type 1 diabetes, it can occur in patients with any form of diabetes. In fact, the Phase II Fremantle Diabetes Study from the UK, with 1,724 hospitalised patients, showed the overall incidence of DKA was 35.6/ 10,000 person-years for Type 1 diabetes; 13.3/10,000 person-years for Type 2 diabetes; 121.5/10,000 person-years for adults with latent autoimmune diabetes; and 446.5/10,000 person-years for secondary diabetes.3
DKA in pregnancy is an obstetrical emergency. Quality data are unavailable regarding the incidence and prevlance of DKA in pregnancy according to subtype of diabetes; however, it appears to complicate 5–10% of all pregnancies with pregestational diabetes, and can also occur in females with gestational diabetes.4-6 Without prompt diagnosis and treatment, DKA can be associated with maternal and fetal mortality. However, with an improved understanding of the disease and appropriate management, the associated maternal–fetal morbidity and mortality have improved over the years.4 It is critical to have a very high index of suspicion for DKA during pregnancy, as the symptoms can be misleading and can present without an extreme increase in plasma glucose concentration.7,8 Furthermore, females may not have a diagnosis of diabetes prior to presenting with DKA.
WHAT IS THE PATHOPHYSIOLOGY OF DIABETIC KETOACIDOSIS?
DKA results from an increased glucagon to insulin ratio, with a concomitant increase in counter-regulatory hormones (cortisol, growth hormone), resulting from an absolute or relative insulin deficiency leading to abnormal carbohydrate, protein, and fat metabolism. Because of hypoinsulinaemia, relative cellular hypoglycaemia ensues, resulting in the stimulation of counter-regulatory response. With increased delivery of gluconeogenic precursors (e.g., alanine and glutamine from protein catabolism, glycerol from lipolysis, lactate from muscle glycogenolysis), hyperglycaemia occurs from enhanced endogenous hepatic glucose production, which may be further compounded by renal gluconeogenesis.2,9 In addition to growth and other counter-regulatory hormones, epinephrine activates hormone-sensitive lipase in adipose tissue, leading to enhanced synthesis of nonesterified fatty acids (a substrate for ketogenesis in the liver). Furthermore, hyperglucagonaemia (and hypoinsulinaemia) activates carnitine acyltransferase 1, the rate-limiting enzyme for ketogenesis, eventually contributing to high anion gap metabolic acidosis.10 Osmotic diuresis and multiple electrolyte disturbances follow, leading to hypovolemia and hypertonicity.2
WHY DOES PREGNANCY PREDISPOSE TO DIABETIC KETOACIDOSIS?
There are several pregnancy-related alterations to the metabolic milieu that prime females (with and without diabetes) for DKA development (Figure 1).
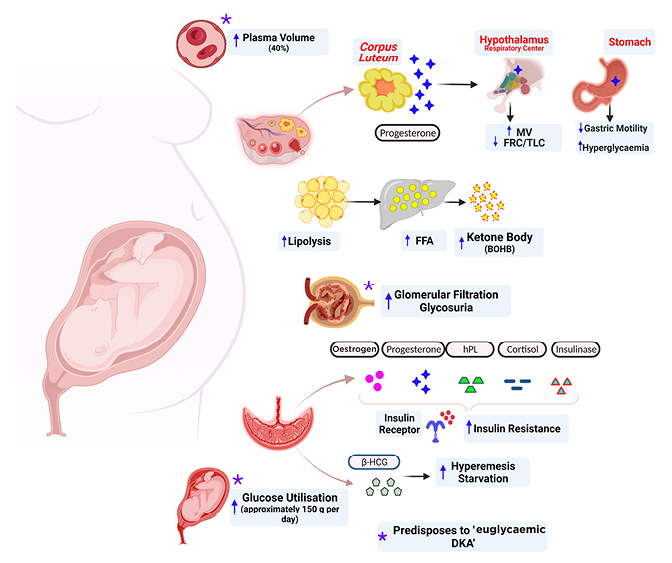
Figure 1: Mechanisms underlying the development of diabetic ketoacidosis in pregnancy.
Created with the assistance of BioRender© 2022.
BOHB: β-hydroxybutyrate; DKA: diabetic ketoacidosis; FFA: free fatty acid; FRC: functional residual capacity; HCG: human chroionic gonadotropin; hPL: human placental lactogen; MV: minute ventalition; TLC: total lung capacity.
Pregnancy is considered an insulin-resistant state whereby insulin sensitivity decreases by 56% by 36 weeks of gestation. Contributors to insulin resistance include increased maternal concentrations of serum oestrogen, progesterone, cortisol, human placental lactogen, TNF-α, and prolactin.11,12
Pregnancy is characterised by fasting hypoglycaemia, with associated hyperinsulinaemia.13 In some cases, elevated human chorionic gonadotropin during early pregnancy is associated with hyperemesis, which could contribute to a relative state of hypoglycaemia, facilitating a state of starvation ketosis.
The fetoplacental unit can utilise up to 150 g per day of glucose to meet the demands of the growing fetus.14 Hence, to meet this increased need, maternal insulin resistance could possibly be a physiological adaptation. Studies have shown enhanced expression of glucose transporter 1, especially in females with gestational diabetes, which increases glucose uptake by the fetoplacental unit.15,16
In addition to the cellular antagonising properties of progesterone to insulin, progesterone decreases gastrointestinal motility, thereby enhancing hyperglycaemia.
Progesterone also increases minute ventilation by 30–50%, leading to a state of respiratory alkalosis, which is potentiated by bibasilar alveolar collapse from the gravid uterus, decreasing both functional residual capacity and total lung capacity by 10–20%.17-20
Renal compensation of respiratory alkalosis through enhanced bicarbonate excretion decreases the buffering capacity during an acidotic state.
Compared with females without diabetes, plasma concentrations of free fatty acids and β-hydroxybutyrate (BOHB) are significantly higher during all trimesters of gestation in females with both pregestational diabetes and gestational diabetes.21,22
Intriguingly, all females who are pregnant are at risk of ketosis, even without underlying diabetes. This was demonstrated in a prospective study comparing females who were pregnant without diabetes who fasted during Ramadan to age-matched non-fasting females who were pregnant. The fasted group were documented to have significantly higher rates of ketosis (11.3% versus 5.0%) and worsening clinical complaints (nausea, vomiting, and dizziness).23
Taken together, these metabolic changes mean that DKA can develop faster and at lower glucose concentrations during pregnancy. In fact, ‘euglycaemic DKA’, broadly defined as plasma blood glucose concentration <14 mmol/L (252 mg/dL), has been well described during pregnancy.24 In addition to the general explanations for the aetiology of DKA in pregnancy (outlined above), additional potential contributors to euglycemic DKA include significantly increased glomerular filtration rate (increased by approximately 60%) during pregnancy with associated glycosuria;14,25 enhanced maternal (oestrogen and progesterone), and fetal (anabolic state) glucose utilisation;25 and a concomitant increase in plasma volume by 40% compared with the pre-pregnancy state, leading to a dilutional effect.25
Hence, all females who are pregnant, regardless of the type of diabetes, are at risk of developing DKA and the underlying pathophysiological mechanisms leading to DKA in different subtypes of diabetes are presumably similar.
WHAT ARE THE RISK FACTORS FOR DIABETIC KETOACIDOSIS IN PREGNANCY?
A 10-year retrospective review of 37 hospitalisations for DKA during pregnancy in a tertiary care centre demonstrated that hyperemesis and β-sympathomimetic drugs were associated with 57% of DKA episodes, with non-compliance and physician management errors occurring in 24%.26 Multiple other studies have documented additional risk factors, including concurrent infections, insulin device failure, missed insulin doses, maternal gastroparesis, and undiagnosed diabetes.27-31 Use of glucocorticoids for fetal lung maturation towards the later part of pregnancy in conjunction with maternal insulin resistance can markedly increase the risk for DKA. Hence, careful monitoring of blood glucose concentration and timely treatment of hyperglycaemia following glucocorticoid administration is warranted.
CLINICAL PRESENTATION OF DIABETIC KETOACIDOSIS
The clinical signs and symptoms associated with DKA can often be misconstrued as normal pregnancy. For example, as nausea and vomiting is associated with >70% of all pregnancies, underlying DKA could potentially be missed if not suspected.32 Common clinical symptoms of DKA in pregnancy include nausea, vomiting, and abdominal pain.4 Patients can also present with generalised fatigue, polyuria, and polydipsia secondary to hyperglycaemia, evidence of volume depletion including dry mucous membranes, increased skin turgor, tachycardia, and hypotension. In severe cases, rapid, shallow breathing (Kussmaul’s respiration) with the classic fruity odour from ketones could be present. Patients with severe DKA can also present with altered mental status, lethargy, and disorientation. Rarely, patients can develop cerebral oedema, causing obtundation and coma. As symptoms are non-specific and females may not have a prior diagnosis of diabetes at presentation, DKA should be considered in any individual who is pregnant and who is unwell.
LABORATORY FINDINGS AND DIAGNOSTIC CRITERIA FOR DIABETIC KETOACIDOSIS
Venous blood collection for plasma glucose, serum electrolytes, renal function (including blood urea nitrogen and serum creatinine), serum osmolality, serum ketones (quantitative or qualitative based on the availability), blood gas, and urine (mid-stream preferably) for a urinalysis and urinary ketones should be collected as part of the initial evaluation.33 The Joint British Diabetes Society’s (JBDS) diagnostic criteria for DKA in pregnancy include positive urinary ketones more than (++) or serum ketones >3.0 mmol/L (in high-risk cases, serum ketones >1.5 mmol/L) and evidence of metabolic acidosis with a venous blood pH <7.30 and/or bicarbonate <15 mmol/L.34 In addition, an anion gap (Na– [Cl+ HCO3]) of >12 mmol/L indicates the presence of an increased anion gap metabolic acidosis.33
While evaluation of ketonaemia via nitroprusside test (serum and urine) is highly sensitive, it helps estimate only acetone and acetoacetate levels but not BOHB, which is elevated in a >10:1 ratio compared with other ketoacids during an insulin-deficient state.33 Hence, quantitative assessment of BOHB is helpful if available. Serum electrolyte concentrations, particularly potassium and phosphate concentrations, should be carefully monitored. They can initially be ‘pseudonormal’ during early stages of DKA due to transcellular shifts (from insulin deficiency) and associated hypertonicity while masking a total body deficit. About 16–25% of patients with DKA can have a non-specific elevation of serum amylase and lipase concentration.35
MANAGEMENT OF DIABETIC KETOACIDOSISIN PREGNANCY
Guidelines for the management of DKA in females who are pregnant are broadly the same as that of an individual who is not pregnant, and is similar for all subtypes of underlying diabetes, including identification and treatment of precipitating factors, fluid replacement, insulin therapy, and electrolyte repletion (Figure 2). In addition, fetal monitoring is warranted to identify any deleterious consequences of maternal acidosis on the fetus. All patients who are pregnant with confirmed or suspected DKA should, therefore, be admitted to highly specialised units (preferably intensive care units) capable of managing DKA and providing continuous fetal monitoring.
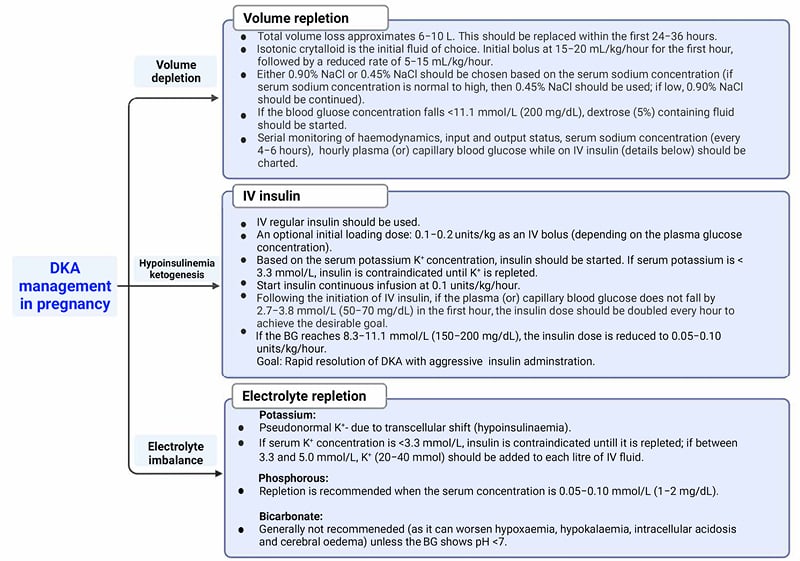
Figure 2: Management of diabetic ketoacidosis during pregnancy.
BG: blood gas; DKA: diabetic ketoacidosis; K+: potassium; IV: intravenous; NaCl; sodium chloride.
Identify and Treat the Underlying Cause
When DKA is confirmed, every attempt should be undertaken to determine the possible precipitating factor(s). A comprehensive history to evaluate for factors such as underlying infection, cardiovascular event, use of glucocorticoids, insulin omission or malfunction of insulin delivery systems, and use of expired insulin should be obtained. An individualised workup should subsequently be completed.
Fluid Management
Osmotic diuresis secondary to hyperglycaemia can lead to significant volume depletion, as high as 10 L. Volume resuscitation improves hyperglycaemia and enhances peripheral insulin action blunted by hyperosmolarity and hyperglycaemia (in patients with normally functioning kidneys).33 One of the primary management goals should be the replacement of total volume loss within the first 24–36 hours of presentation. Isotonic fluid, 0.9% sodium chloride (NaCl), is the recommended initial crystalloid but can be guided by the haemodynamic status on presentation.4,34 About 50% of the fluid should be replaced within 24 hours of presentation. A widely followed protocol is to start isotonic crystalloids at the rate of 15–20 mL/kg/hour (approximately 1.0–1.5 L for a 70 kg adult) for the first hour, followed by a decreased rate of 5–15 mL/kg/hour. The decision to continue 0.90% NaCl versus 0.45% NaCl should be made based on serum sodium trends (rapid improvement of corrected serum sodium can increase the risk for cerebral edema), serum osmolality, and urine output.36 For example, following initial resuscitation, 0.45% NaCl could be substituted at the rate of 250–500 mL/hour if the corrected serum sodium is normal or elevated; whereas, if low, 0.90% NaCl should be continued with serial monitoring of sodium. The total fluid requirement may be relatively lower compared with an individual who is not pregnant.34
Furthermore, individuals with kidney disease, heart failure, or pre-eclampsia may need a more conservative approach to fluid replacement, which should be individualised according to initial tolerance and overall clinical status. Once the blood glucose concentration reaches 11.1 mmol/L (200 mg/dL), dextrose containing fluids (5% or 10% dextrose) should be initiated to prevent hypoglycaemia while continuing intravenous (IV) insulin therapy, as the goal is to inhibit ongoing ketogenesis that was driven by hypoinsulinaemia.4 Successful fluid resuscitation is reflected by improvement in volume status, haemodynamics, serum sodium concentration, blood glucose concentration, and overall clinical status.
Intravenous Insulin
Continuous IV insulin infusion therapy is the mainstay of DKA management to correct the underlying metabolic derangement. The goal of IV insulin therapy is not only to control hyperglycaemia (by promoting glucose uptake at the periphery and inhibiting gluconeogenesis) but also to suppress ongoing ketogenesis. Insulin should be initiated based on serum potassium concentrations, along with fluid resuscitation. Adequate initial volume resuscitation prior to initiation of insulin therapy could potentially enhance insulin sensitivity and decrease insulin requirement. The initial loading dose of 0.1–0.2 units/kg as an IV bolus is endorsed by the American College of Obstetricians and Gynecologists (ACOG).4 However, the JDBS recommends starting fixed-rate insulin at 0.1 unit/kg/hour.34 Studies outside of pregnancy have shown no difference in the rate of plasma glucose concentration or anion gap improvement, the incidence of hypoglycaemia, length of hospital stay, and the overall outcome of DKA when treated using continuous insulin infusion therapy, with or without the initial IV bolus.36-38
A continuous IV insulin infusion at the rate of 0.1 unit/kg/hour should be initiated (with or without the initial bolus) only if the serum potassium concentration is >3.3 mmol/L, and should be delayed until hypokalaemia is appropriately corrected. Plasma or capillary blood glucose concentration should be assessed on an hourly basis. If the plasma/capillary blood glucose concentration does not fall by at least 2.7–3.8 mmol/L (50–70 mg/dL) in the first hour, the insulin infusion rate must be doubled, and this titration of insulin should be carried out every hour until a steady-state blood glucose concentration is achieved. Once the plasma/capillary blood glucose concentration reaches 11.1 mmol/L (200 mg/dL), in addition to dextrose-containing fluids (to avoid hypoglycaemia), the insulin infusion dose needs to be reduced to 0.05–0.10 unit/kg/hour. The eventual goal is to maintain a blood glucose concentration between 5.6–8.3 mmol/L (100–150 mg/dL), with the smallest insulin dose possible, until complete inhibition of ketogenesis is achieved, as evidenced by the normalisation of the anion gap (if normal renal function).4 While IV insulin is ongoing, periodic monitoring and replacement of electrolytes, especially potassium, is essential. Temporary discontinuation of insulin may be needed to facilitate potassium repletion.
Once the anion gap normalises, the transition from continuous IV insulin to subcutaneous insulin with a 2–4 hour overlap is made, primarily to prevent rebound hyperglycaemia. However, it is reasonable to continue the long-acting insulin throughout DKA treatment if the patient is on a known multiple daily injection insulin regimen.34 This practice can reduce the risk of rebound hyperglycaemia and recurrent DKA, which can occur with premature discontinuation of IV insulin and/or inadequate overlapping with subcutaneous insulin. If the patient was on an insulin pump, it should be restarted following DKA resolution.
Electrolyte Replacement
Hypoinsulinaemia, hyperosmolarity, and acidosis associated with DKA cause total body potassium deficit despite normal serum potassium concentration (due to transcellular shift). Therefore, rapid correction of DKA could potentially increase the risk of hypokalaemia and life-threatening cardiac arrhythmias, warranting close monitoring of serum potassium. If the initial serum potassium concentration is <3.3 mmol/L (as opposed to >5.5 mmol/L), IV insulin therapy is contraindicated until potassium is adequately repleted. However, fluid resuscitation should be initiated regardless of the serum potassium concentration. Certainly, potassium could be added to the intravenous fluid simultaneously to ensure that serum potassium is maintained within the acceptable range of 3.3–5.5 mmol/L.4
Hypophosphataemia can also ensue with aggressive fluid hydration and insulin treatment due to transcellular shift. Usually, severe hypophosphataemia is rare, and replacement is indicated when the concentration is 0.05–0.11 mmol/L (1.00–2.00 mg/dL). Rapid or over-correction of phosphate can precipitate hypocalcaemia and, hence, careful monitoring of serum calcium is warranted.39,40
The use of bicarbonate in patients with DKA is controversial as it increases the risk of hypoxaemia, hypokalaemia, intracellular acidosis, and cerebral oedema.33,36 Bicarbonate treatment is not recommended unless the patient has severe metabolic acidosis with a pH <7.0 and should be done under expert supervision.
MONITORING RESPONSE TO DIABETIC KETOACIDOSISTREATMENT
During DKA treatment, patients’ capillary blood glucose should be monitored hourly while on IV insulin therapy, and serum electrolytes for potassium, phosphorous, and calcium concentration and anion gap every 4 hours. The JDBS endorses monitoring venous bicarbonate and potassium at 1 hour, 2 hours, and then every 4 hours.41 However, monitoring serum bicarbonate may be helpful only during the first 6 hours following the initiation of the DKA protocol because of increased incidence of hyperchloraemic metabolic acidosis (normal anion gap) secondary to administration of isotonic crystalloids (NaCl) for volume resuscitation.33 Clearance of urinary ketone lags behind the clearance from the blood and, hence, monitoring serum anion gap is advisable (when renal function is normal). The recommended metabolic targets to achieve during DKA treatment include an increase in venous bicarbonate concentration by 3.0 mmol/L/hour; a decrease in capillary blood glucose level by 3.0 mmol/L/hour (54 mg/dL/hour); and a decrease in blood ketone concentration by 0.5 mmol/L/hour.34 Strict criteria to determine DKA resolution in pregnancy are not established; however, one can generally consider biochemical resolution if the following parameters are satisfied: serum ketone concentration <0.6mmol/L; calculated anion gap ≤12.0 mmol/L; venous pH >7.3; and serum bicarbonate concentration >15.0 mmol/L.33,42
FETAL MONITORING DURING DIABETIC KETOACIDOSIS
Severity of maternal DKA determines the severity and frequency of fetal heart rate changes. However, in most cases, these changes are reversible following the correction of maternal acidosis.43 Continuous fetal monitoring is recommended if the pregnancy is >24 weeks of gestation during the DKA episode.44 Initial fetal heart rate tracing during the DKA episode can show decreased or absent variability, absent accelerations, or late decelerations. In addition, Doppler ultrasound studies using pulsatility index have demonstrated transient alteration in blood flow, particularly redistribution of blood flow in the middle cerebral (reduced) and umbilical arteries (increased).43,45 There are no clear recommendations for the duration of continuous fetal monitoring; however, in most centres, it is continued until the resolution of DKA with frequent monitoring post resolution. Typically, DKA is not an inidication for delivery but this decision should be made on a case-by-case basis.
WHAT ARE THE OUTCOMES FOLLOWING DIABETIC KETOACIDOSIS IN PREGNANCY?
The maternal and fetal outcomes reported in previously published retrospective studies and case series of DKA in pregnancy are outlined in Table 1.7,14,24,29,46-51. Original studies were chosen through literature search in the PubMed database using combinations of the following key search terms: “diabetes”, “gestational diabetes”, “GDM”, “Type 2 diabetes”, “hyperglycemia”, “ketoacidosis”, “pregnancy”. The reference list of included articles were also examined to identify articles not retrieved using this search strategy. Case reports and case series with less than five patients were not included in this review. There were no conveivable differences nor any detailed reports about maternal and fetal outcomes following the DKA events in different diabetes category.
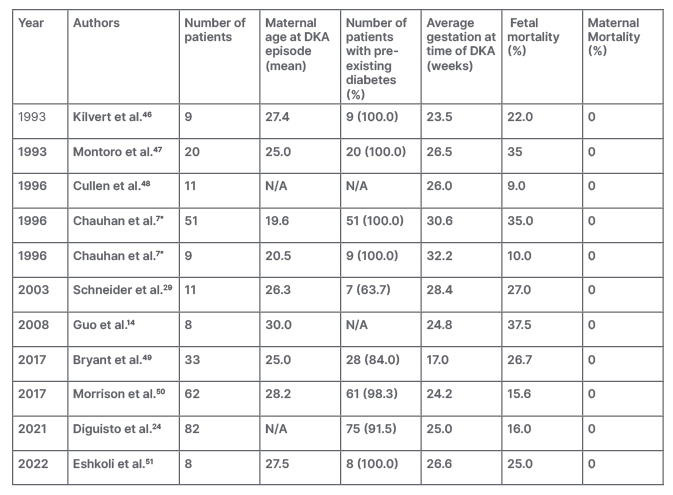
Table 1: Prior published studies of diabetic ketoacidosis in pregnancy (case reports excluded).
*Two historical cohorts are reported in this study: Cohort 1 (n=51) from 1976 to 1981 and Cohort 2 (n=9) from 1986 to 1991.
DKA: diabetic ketoacidosis; N/A: not available.
Maternal Outcomes
The overall maternal mortality associated with DKA is <1%. However, similar to individuals who are not pregnant, DKA can be associated with significant morbidity, including hypotension (from severe dehydration), organ dysfunction (from acidosis), and cardiac dysrhythmias (from electrolyte abnormalities, including hypokalaemia) in a female who is pregnant. All these metabolic changes could potentially be prevented and/or reversed following the prompt initiation of treatment. Cerebral oedema with neurological symptoms is a rare but serious complication of severe DKA (in the population that is not pregnant) with a >70% mortality rate (particularly encountered in children).2
Fetal Outcomes
The reported fetal mortality ranges from 9–36%.7,46-48,52 Various hypotheses have been postulated outlining the pathophysiology of fetal demise. Both ketones and glucose cross freely across the placenta.53 Fetal hyperglycaemia due to maternal hyperglycaemia leads to fetal osmotic diuresis. Furthermore, fetal acidaemia, from transfer of ketoacids across the placenta, can lead to vasoconstriction of the uteroplacental blood vessels. In addition, a decrease in 2,3 bisphosphoglycerate (decreased phosphate) could decrease the fetal oxygen delivery (leftward shift of maternal oxy-hemoglobin curve with increased oxygen affinity). Fetal cardiac arrhythmias can develop in the setting of electrolyte disturbances, particularly hypokalaemia. Despite the deleterious consequences, all these adverse outcomes can be prevented or even reversed with proper treatment of DKA. In addition to the immediate sequelae of DKA, data have associated ketone body exposure with a negative impact on long-term neurodevelopmental abnormalities, particularly intelligence and psychomotor development.54-56 However, the biological mechanism is not clear.
DIABETIC KETOACIDOSIS PREVENTION
The measures to prevent maternal and fetal adverse outcomes due to DKA should begin during preconception counseling. It is imperative to prepare a female with diabetes for pregnancy through adequate education about the importance of diabetes control prior to pregnancy. Close monitoring and regular insulin titration are critical during pregnancy. In addition, females should receive education on precipitating factors, signs, and symptoms of DKA, home monitoring of urine ketones, and how to seek help on time. A multidisciplinary team involving an endocrinologist (trained in managing diabetes in pregnancy), high-risk obstetrician, maternal-fetal medicine specialist, and diabetes educators should ideally be involved in caring for a females who are pregnant with diabetes.

Box 1: Clinical Pearls: Diabetic ketoacidosis in pregnancy.
DKA: diabetic ketoacidosis.
CONCLUSION
DKA in pregnancy is an obstetrical emergency associated with poor pregnancy outcomes. Compared with an individual who is not pregnant, a female who is pregnant with diabetes is at higher risk of developing DKA, and the condition can occur with lesser degrees of hyperglycaemia. DKA can also be the first presentation of diabetes in pregnancy. A high index of suspicion along with prompt diagnosis and management are critical to ensure optimal outcomes (Box 1). Further studies are warranted to develop improved strategies to prevent DKA during pregnancy and clarify the long-term sequelae of ketosis on offspring of females with DKA.