Abstract
Gastrointestinal (GI) symptoms represent an important and often poorly appreciated reason of morbidity in diabetes mellitus. Diabetes can affect nearly all parts of the GI tract; however, data on the prevalence of ‘diabetic gastroenteropathy’ are inconsistent. The significance of disturbed GI motility in diabetes across the patient spectrum and pathophysiological basis also remain inadequately defined. Fluctuating glucose levels, altered drug pharmacokinetics, variable absorption of nutrients, and impaired quality of life are important consequences of GI dysfunction. Diabetic gastroparesis is the best characterised manifestation of GI motility disorder in diabetes. Since there is a poor correlation between subjective GI symptoms and objective motility findings, a diagnosis of delayed emptying in diabetes requires a proper measurement of gastric emptying. There are fewer studies on intestinal motility in diabetes than those on the stomach. Several established modalities exist for the assessment of gastroenteropathy but the lack of standardisation, exposure to radiation, advanced data interpretation, and high cost limit their widespread use. While existing therapeutic choices for the management of diabetic gastroenteropathy are suboptimal, many potential novel agents are in progress. Both endocrinology and gastroenterology specialties working together will facilitate screening and treating patients with diabetes and GI dysmotility.
INTRODUCTION
People with Type 1 and Type 2 diabetes mellitus (T1 and T2DM) can present with a diverse range of symptoms in all levels of the gastrointestinal (GI) tract. However, there are inconsistent data on the prevalence of GI symptoms, and the frequency of symptoms is much higher when data are reported by a gastroenterologist than reported by a diabetologist.1 The picture is further confounded by the dissociation between GI symptoms and the transit profile. Although motility disorders in DM are pan-enteric, perhaps the best-known diabetic GI complication is gastroparesis (Gp), or abnormally delayed gastric emptying (GE).2 GI dysfunctions in diabetes not only have a detrimental effect on the quality of life, but also significant medical consequences. In recent years, the data in regard to the underlying pathophysiology of diabetic gastroenteropathy is expanding.3 The importance of the evaluation of the entire GI tract in patients with diabetes and motility impairment is also being increasingly recognised. In this context, this review aims to explore the GI motility disorders in diabetes, focusing on the pathophysiology, effects on glycaemia, limitations of assessment methods, unmet needs in the treatment, and an outlook on future research.
PATHOPHYSIOLOGY
Understanding the pathophysiology of DM-induced GI dysmotility is important to develop therapies to correct or prevent the underlying mechanism of this widely prevalent disorder. DM-induced GI dysmotility is multifactorial and not completely elucidated. It can affect any part of the GI tract via a composite of several dysfunctional factors.4 Autonomic neuropathy and hyperglycaemia are the two main factors implicated in the pathogenesis of DM-induced GI dysmotility.5 Parasympathetic nerve dysfunction, known as autovagotomy, in DM leads to gastric stasis and rapid small bowel transit. Sympathetic nervous system dysfunction due to loss of α2 adrenergic tone causes small bowel dysmotility, abnormal fluid transport, and nocturnal faecal incontinence due to the loss of internal anal sphincter tone.6 Prolonged hyperglycaemia alters GE, myoelectrical activity, and gastrocolic reflex.6
Although studies have shown an association between poor glycaemic control or autonomic neuropathy and GI symptoms in DM, these symptoms can develop before the onset of autonomic neuropathy or have a poor correlation with neuropathy. Hence, other pathophysiological mechanisms are likely to be present (Figure 1). These mechanisms have been studied in human and experimental models of DM, which include enteric myopathy and neuropathy.7 Atrophy of smooth muscles and apoptosis of neurons have been observed in experimental models of DM as a result of autoimmunity and metabolic derangements leading to alteration of critical cellular pathways (e.g., phosphatidylinositol 3-kinase pathway) and signalling of trophic factors. Reduction of insulin or insulin-like growth factor 1 signalling in DM results in atrophy of intestinal smooth muscles. This leads to the decreased production of trophic factors such as stem cell factor, which results in loss of pacemaker interstitial cells of Cajal (ICC) or trans-differentiation into a smooth muscle phenotype.8 Trans-differentiation also leads to an imbalance in number of excitatory and inhibitory enteric neurons and neuropeptides (e.g., vasoactive intestinal peptide, nitric oxide, calcitonin gene-related peptide, substance P).9 Loss of ICC in myenteric plexus leads to obliteration of slow-phase peristaltic movements and gastric dysmotility. ICC located in the muscle layer impair neurotransmission in the enteric and autonomic nervous system, as well as to smooth muscle.3 Moreover, abnormal central processing of visceral pain has been reported in DM.
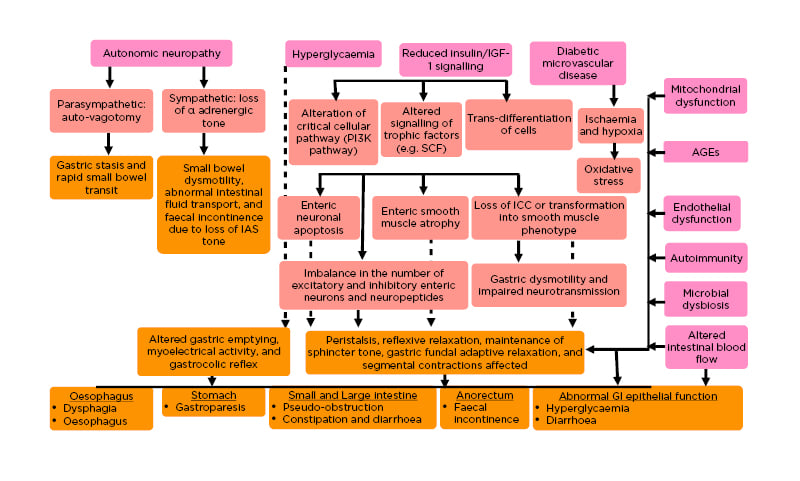
Figure 1: Pathogenesis of diabetes mellitus-associated gastrointestinal dysmotility.
Key pathogenetic factors and their effects are shown in pink and blush red-coloured boxes, respectively. Consequent clinical effects on various parts of the GI tract are depicted in dark yellow boxes.
AGE: advanced glycation end product; GI: gastrointestinal; IAS: internal anal sphincter; ICC: interstitial cells of Cajal; IGF-1: insulin-like growth factor 1; PI3K: phosphatidylinositol 3-kinase; SCF: stem cell factor.
Diabetic microvascular disease causing ischaemia and hypoxia, leading to oxidative stress; mitochondrial dysfunction; advanced glycation end products; and endothelial dysfunction mediated by peroxynitrite are other mechanisms for DM-induced dysmotility.3
Complex intestinal motor functions such as peristalsis, reflexive relaxation, maintenance of sphincter tone, gastric fundal adaptive relaxation, segmental contractions, and also intestinal blood flow are altered in DM due to the collective effect of various defective factors, discussed above, such as autonomic neuropathy, loss of ICC, and imbalance of enteric neurotransmission.3 These altered GI motor functions lead to dysphagia and reflux oesophagitis, Gp, intestinal pseudo-obstruction, alternating constipation and diarrhoea, and faecal incontinence due to anal sphincter dysfunction. Abnormal GI epithelial function due to defective signalling of trophic factors and enteric neuropathy lead to enhanced nutrient transport and consequent hyperglycaemia, whereas abnormal intestinal transport of salt and water leads to diabetic diarrhoea. Compromised intestinal vascular flow in DM can lead to intestinal mucosal dysfunction, which can indirectly affect motility.4
EPIDEMIOLOGY
The prevalence of GI motility disorders in diabetes varies depending on the place of study (tertiary centre versus community-based), the definition employed (self-report versus validated questionnaire versus ecological momentary assessment), and the specialty involved (gastroenterology versus diabetology).10 The risk factors include older age and longer duration of diabetes, female sex, higher HbA1c level, lower socio-economic status, greater prevalence of microvascular complications (particularly neuropathy), anti-diabetic medication use (metformin and acarbose), and associated depression.11 Among oesophageal symptoms, reflux is seen in up to 24% of patients with T1DM and 60% of patients with T2DM, but dysphagia is less common (4–13% in different studies).10 Recent analysis from the follow-up cohort of Diabetes Control and Complications Trial (DCCT) showed that 47% of patients with T1DM have delayed GE of a solid meal.12 If diabetes is long-standing and poorly controlled, the prevalence of Gp in T1 and T2DM is likely comparable. The data from Mayo Clinic, Scottsdale, Arizona, USA, provided the cumulative incidence of Gp (defined by a scintigraphic study and/or symptoms suggestive of Gp). Over the course of 10 years, 5% of people with T1DM and 1% of those with T2DM developed Gp.13 The upper abdominal symptoms (nausea, bloating, early satiety, or upper abdominal pain) range between 10% and 40% in different studies.10 However, the GE data with T2DM is scarce. Furthermore, symptoms often do not correlate with GE. Interestingly, some scintigraphy data are demonstrating GE is relatively more rapid in people with well-controlled T2DM.14 The prevalence of diarrhoea is seen in up to 41% of patients with T1DM and 35% of patients with T2DM, while constipation is seen in up to 33% of patients with T1DM and up to 28% of patients with T2DM in different trials.10 In the National Health and Nutrition Examination Survey (NHANES) dataset, which evaluated the prevalence of GI disturbances through the Bowel Health Questionnaire, after adjusting the covariates, chronic diarrhoea was more prevalent in patients with diabetes than in those who do not have diabetes, whereas chronic constipation (CC) was not.15
CLINICAL PRESENTATIONS
The prevalence of GI symptoms in patients with diabetes is higher than in the general population. Clinical manifestations of GI motility disorders can be classified into three sections according to the site of involvement.
Oesophagus
Gastro-oesophageal reflux and dysphagia are two common oesophageal motility disorders. Reflux disorders are more prevalent and mostly asymptomatic but can present with heartburn or cough.16
Stomach
Gastric symptoms are mostly related to slow GE called Gp. Post-prandial fullness, early satiety, bloating, nausea, vomiting, and upper abdominal pain are the common presentations.17 Vomiting and early satiety are more frequent in diabetic gastroparesis (DGp), whereas abdominal pain is more frequent in idiopathic Gp.18 Symptoms are more common in women, patients who are obese, and those with coexistent depression.19 Interestingly, new symptoms sometimes appear and old symptoms disappear, with total prevalence remaining constant. This symptom ‘turnover’ may be as high as 15–25% over 2 years.20 Sometimes Gp may present with poor nutritional status or unusual changes in post-prandial glycaemic patterns, such as erratic peaks and troughs in glucose concentrations.
Intestine
Symptoms at the intestinal level are constipation, diarrhoea, pain, and bloating. Among these, CC is the most commonly reported. DD is painless, chronic (>6 weeks), watery diarrhoea.21 Nocturnal diarrhoea and faecal incontinence are two of the most typical findings of DD. Slow intestinal transit may predispose to small intestinal bacterial overgrowth, which can also lead to diarrhoea.22
DIAGNOSTIC APPROACH FOR GASTROINTESTINAL MOTILITY DISORDERS IN DIABETES
The symptoms of DM-induced intestinal dysmotility can be diverse as it can affect any part of the GI tract as outlined above. None of the symptoms are specific for DM-induced dysmotility. Organic diseases such as gallstone disease, gastro-oesophageal reflux disease, GI malignancies, and autoimmune conditions like coeliac disease can be associated with DM (commonly with T1DM) and mimic symptoms of DM-induced GI dysmotility. Among neuroendocrine tumours, glucagonoma can present with glucose intolerance, diarrhoea, and abdominal pain. Hence, it is important to distinguish GI motility disorders in diabetes from other organic diseases.23 A review of anti-diabetic medications is also important since agents like metformin and acarbose can cause nausea, vomiting, flatulence, and diarrhoea, mimicking symptoms of GI dysmotility.
The first step to evaluate symptoms of GI dysmotility in patients with DM is to perform basic laboratory investigations, imaging, and an endoscopy to rule out other organic diseases (Figure 2). GI function testing is recommended if these investigations are non-contributory and symptoms do not respond to symptomatic treatment such as laxatives for constipation. However, the most readily available GI motility tests cannot prove causation by DM-induced dysmotility or gauge the relative contribution of DM in the case of multifactorial causation of dysmotility.23
Investigations that are specific for DM-induced gastroenteropathy like pancreatic polypeptide (a reduction in pancreatic polypeptide is specific) and antro-duodenal motility testing (Phase II and post-prandial hypomotility and increased Phase III motility, specific for DGp) are not readily available.24,25 Other organ involvement (e.g., cardiac autonomic neuropathy) increases the probability of DM-induced GI changes. Commonly available tests like high-resolution manometry showing oesophageal hypomotility and GE studies show that Gp cannot differentiate DM-induced dysmotility from other causes. However, the GE study is important and should be done in patients with poorly controlled diabetes early as it has important implications in glycaemic control.25 Both impaired gastric motility and rapid GE (in up to 20% with impaired GE) can occur, leading to dumping syndrome, which can cause similar symptoms (e.g., unexplained nausea, vomiting) to be differentiated based on GE.26 A detailed approach to DM-induced GI dysmotility is illustrated in Figure 2.
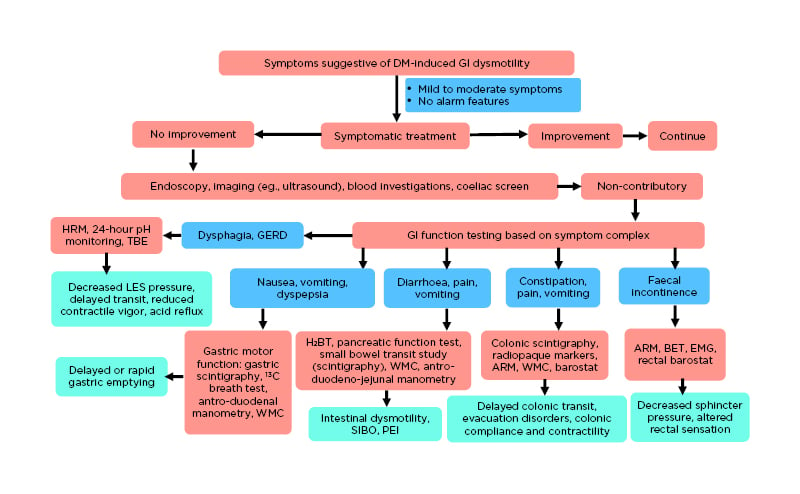
Figure 2: Clinical approach in suspected diabetes mellitus-associated gastrointestinal dysmotility.
ARM: anorectal manometry; BET: balloon expulsions test; EMG: electromyography; GERD: gastroesophageal reflux disease; HRM: high-resolution manometry; H2BT: hydrogen breath test; LES: lower oesophageal sphincter; PEI: pancreatic exocrine insufficiency; SIBO: small intestinal bacterial overgrowth; TBE: timed barium oesophagogram; WMC: wireless motility capsule.
Newer emerging diagnostic modalities include13C-breath test (for GE test based on hepatic metabolism after intestinal absorption, good correlation with scintigraphy), wireless motility capsule (to assess the entire gut and measure regional transit time in a single test without any radiation), and electromagnetic capsules (Motilis 3D-Transit system [Molitis Medica SA, Lausanne, Switzerland] to reflect gut contractility and regional transit time).25,27,28
CLINICAL MANAGEMENT
Management of Diabetic Gastroparesis
Advances in dietary recommendations
The first step in the management of Gp is dietary modification. Patients with DGp tend to have a lower than recommended caloric intake as well as a significant deficiency of micronutrients.29 Multiple small meals (≥6 /day) are preferable than fewer, large ones. It appears logical to avoid hard-to-digest solids and fats, consume larger calorie proportions as a liquid rather than solid, and take solids of small particle size, but this has a limited evidence base.30 Intake of small-particle, low-fat, and low-fibre diets with sufficient hydration may improve GE. Solid foods that are high in fat are probably the offenders. In practice, it has been observed that fat-containing liquids are mostly well-tolerated.31 Smoking and alcohol consumption should be avoided. For individuals who fail to meet their nutritional requirements consistently or regain the lost weight, enteral nutrition is recommended to bypass the dysfunctional stomach.31 In patients with Gp, using parenteral nutrition should be the exception.
Glycaemic control and gastric emptying
The connection between glycaemic control and Gp is not completely understood and may be bi-directional. Hyperglycaemia can delay GE, whereas disturbances of GE affects glycaemic control.32 Data regarding the long-term effect of glycaemic control on GE are conflicting, with several earlier studies finding no correlation in T1DM and T2DM patients.33-35 However, in an evaluation of a subset of the DCCT, the Epidemiology of Diabetes Interventions and Complications (EDIC) cohort, GE weakly related to both HbA1c at the entry into DCCT and the mean HbA1c over the intervening years.12 Another recent retrospective review of patients (both T1 and T2DM) who underwent GE scintigraphy found a significant association of higher HbA1c levels with higher gastric retention at 4 hours.36
There is evidence, although inconclusive, that improvement of glycaemic control can correct abnormally delayed GE in DM, but that an exact threshold of good control may be required. An abnormally slow GE may predispose to hypoglycaemia. The unexplained hypoglycaemic episodes, mainly early in the post-prandial period, may be the sole presenting feature of DGp and warrant prompt evaluation of GE.37
Optimising glycaemia in patients with gastroparesis
Optimisation of glycaemic control is essential to reduce the acute symptoms of Gp, improve nutrient utilisation, and prevent catabolism.38 A survey of patients with DGp revealed an observation that blood glucose control had become more difficult since the diagnosis of stomach dysfunction, with recurrent episodes of both hypo- and hyperglycaemia.39 Delayed GE affects the pharmacokinetics of oral antidiabetics; hence, these agents do not appear to be suitable for effective glycaemic control in patients with T2DM and clinically significant Gp. Patients with T1DM and most of the patients with T2DM and DGp will require insulin for glycaemic control. Compared to basal insulin, the challenges are more complex for the bolus insulin. There are two practical recommendations regarding the bolus insulin for patients with Gp: the use of regular insulin (rather than insulin analogues) and insulin administration after a meal. It is advocated to use continuous subcutaneous insulin infusion (CSII) together with continuous glucose monitoring to improve glycaemic control in patients with Gp.40 Administering of a second wave of prandial insulin through the dual wave CSII could be especially helpful when the carbohydrate emptying is delayed.41 Nevertheless, there is a lack of randomised clinical trials of CSII in DGp. Glucagon-like peptide-1 receptor agonists can exacerbate symptoms of delayed GE and should be avoided.42 Dipeptidyl peptidase-4 inhibitors, contrary to glucagon-like peptide-1 receptor agonists, are unlikely to have a considerable impact on GE.
Current medications: unmet needs
Theoretically, the symptoms of Gp should best be treated with promotility agents that accelerate GE, which should improve symptoms. Nevertheless, there is a poor correlation of symptoms with GE, and enhancement of GE may not provide improvements in symptoms. For DGp, studies with prokinetic drugs have reported improvement in GE, without consistent effects on symptoms or glucose control.43 A recent systematic review demonstrated a relationship if studies using ‘suboptimal’ techniques for assessing GE were removed, but this analysis excluded the motilin receptor agonists. This is further confounded by the fact that certain prokinetic agents have antiemetic properties as well.44
Prokinetics currently in clinical use are metoclopramide and erythromycin. Though each has been shown to improve GE and reduce symptoms, there are problems with these agents (Table 1).45 Consequently, there continues to be a considerable unmet need for patients with Gp. Newer agents including dopamine receptor antagonists and ghrelin, motilin, and 5-HT4 receptor agonists are being investigated, which demonstrate efficacy and have fewer adverse effects (Table 1).
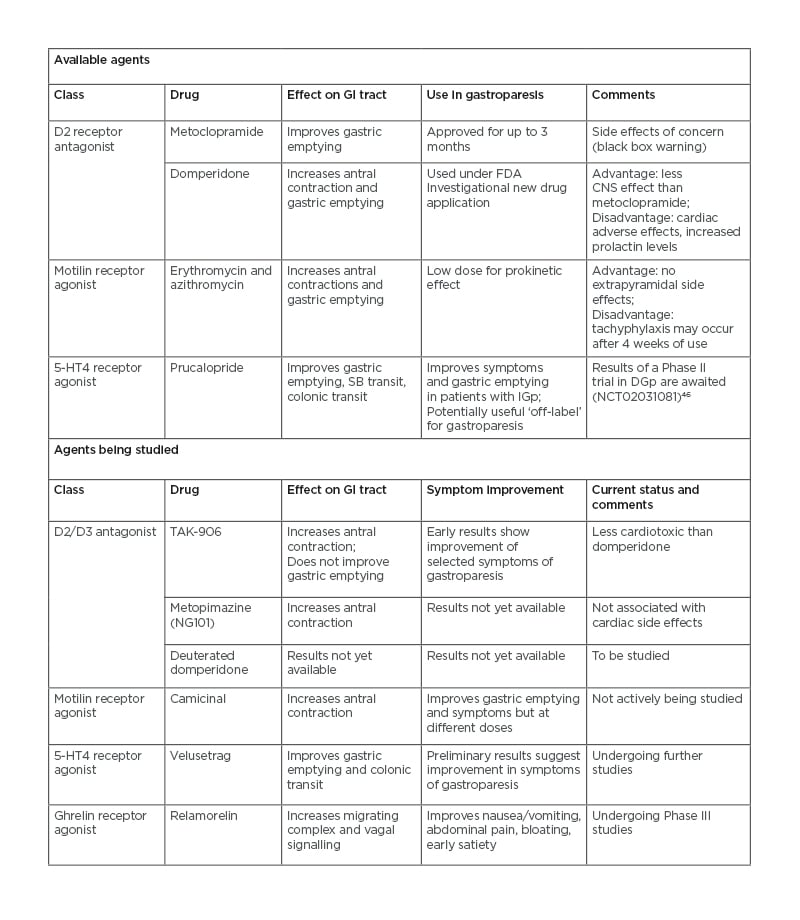
Table 1: Prokinetic agents for gastroparesis that are currently available and under study.
CNS: central nervous system; DGp: diabetic gastroparesis; D2: dopamine type 2 receptor; FDA: U.S. Food and Drug Administration; GI: gastrointestinal; IGp: idiopathic gastroparesis; SB: small bowel; 5-HT4: 5-hydroxy tryptamine receptor 4.
Treatment for refractory gastroparesis
For resistant Gp, which is not responding to dietary modifications and pharmacotherapy, surgical options such as gastric electric stimulation (GES), pyloric surgery (PS) such as a pyloromyotomy or pyloroplasty, or a combination (GES+PS) may be considered. Recent studies have shown that combined GES+PS and GES improve nausea and vomiting better than PS alone, particularly in DGp compared with idiopathic Gp.47 GES gastric pacing delivers high frequency (12 /minute) low-energy pulses via a pacemaker to gastric serosa along the greater curvature. GES is recommended for use in DGp by the U.S. Food and Drug Administration (FDA). Accelerated GE, improved gastric accommodation, and central effects (via the vagus nerve) mediate the beneficial effects of GES. Infection is the major limiting side effect (10%), requiring treatment discontinuation.48
Several relatively non-invasive endoscopic options are upcoming for refractory Gp. Intra-pyloric botulinum toxin injection can be used for Gp in uncontrolled studies. However, placebo-controlled studies have failed to show a benefit.49 Data from a multicentre, non-randomised study of 30 patients with refractory Gp have shown efficacy and technical feasibility of gastric per-oral endoscopic myotomy (G-POEM) with pyloromyotomy (a natural orifice endoscopic transluminal endoscopic surgery; NOTES) leading to normalisation of GE time.50 In a prospective matched cohort study comparing G-POEM and laparoscopic pyloroplasty, G-POEM had significantly lower post-operative morbidity with comparable improvement in GE and symptoms.51 Hence, there is a current trend of shifting from surgical management of refractory Gp to less morbid endoscopic procedures.
Management of Diabetic Enteropathy
Diabetic diarrhoea
Initial treatment should be directed towards the correction of fluid and electrolyte imbalances, with implementation of measures to optimise glycaemic control. Two of the most frequently prescribed anti-diarrhoeals are loperamide and diphenoxylate. However, for DD their use is off-label.21 Unlike diphenoxylate, loperamide is peripherally acting and, thus, is preferred. In DD, bile acid-binding resins may be of therapeutic value, and these agents also reduce HbA1c levels.21 The supportive evidence for use in DD is limited to case reports only. One of the best-studied medications for DD is clonidine.52 However, its use is limited largely by hypotension, mostly in patients with postural hypotension secondary to autonomic neuropathy. In refractory cases of DD, long-acting somatostatin analogues (e.g., parenteral octreotide) may be considered.53 However, high cost, steatorrhoea, gallstones, and dysglycaemia are the limiting factors.
Eluxadoline is one promising future drug for the treatment of DD. Currently, it is a μ-opioid agonist, δ-opioid receptor antagonist, and κ-opioid receptor agonist approved for diarrhoea-predominant irritable bowel syndrome.54 Mixed opioid receptor agents are associated with less constipation and have low potential for dependence or tolerance as compared to their counterparts (μ-receptor agonist). A combined Phase II/III study of eluxadoline for the treatment of DD is currently underway.
Diabetes-associated chronic constipation
Treatment intends to have an improvement of symptoms and restoration of bowel function by accelerating colonic transit and facilitating defecation.55 Implementation of lifestyle and dietary modifications should be done prior to opting for prescription medications. A high-fibre diet, adequate water intake, and physical activity are regularly recommended. However, increasing dietary fibre appears to be useful in those with a deficiency in fibres and too much fibre intake can exacerbate bloating and flatulence.56 High-fibre diets fail to improve bowel movements in people with slow transit or defecation problems.57
If satisfactory relief is not obtained with dietary modifications, the standard treatment is the use of laxatives. However, no studies have assessed a stepwise approach to laxative therapy. The Asian Neurogastroenterology and Motility Association (ANMA) recommends that treatment should start with bulk-forming laxatives, followed by an osmotic laxative and stimulant laxative in individuals who are not responding to bulking agents.58 Lactulose appears to be a suitable and effective osmotic laxative for managing CC in patients with diabetes.59 Nevertheless, well-designed, placebo-controlled clinical trials of available laxatives are limited.
Newer agents such as the 5-HT4 agonist and chloride channel activators can be considered for resistant cases. A novel 5-HT4 agonist, prucalopride, has been approved for CC.60 Lubiprostone, a chloride channel activator, is found to be a safe and effective treatment option for diabetes-related CC.61
CONCLUSION AND FUTURE DIRECTIONS
DM-induced GI dysmotility is common and can affect any part of the GI tract, causing significant disability. However, treatment of diabetic patients with GI dysmotility is limited and currently includes tight glycaemic control and symptom-based management in close consultation with both endocrinologists and gastroenterologists. However, such therapy can be ineffective in a significant subset of patients due to irreversible or unidentified underlying disease mechanisms. The current understanding of mechanisms of DM-induced GI dysmotility is still inadequate, although it has been evolving rapidly in the last decade. Mechanisms other than autonomic neuropathy and hyperglycaemia help to explain why GI symptoms often precede or have a poor correlation with diabetic autonomic neuropathy. DM leads to an altered microenvironment of the enteric nervous system and ICC, leading to either apoptosis or trans-differentiation. Oxidative stress from diabetic microvascular disease, microbial dysbiosis, mitochondrial dysfunction, autoimmune mechanisms, reduction in trophic factor signalling (insulin or insulin-like growth factor 1 pathway), alteration of key cellular pathways, and post-transcriptional regulation of protein synthesis by micro-RNAs can lead to this altered microenvironment. Identification of reversible risk factors holds the key to newer treatment modalities. For example, restoring the trophic signals could restore trans-differentiated ICC and stem cell therapies can reverse ICC apoptosis, leading to resolutions of GI symptoms.