Abstract
Diabetes is defined as chronic hyperglycaemia due to insufficient insulin action. Over the last few decades, various different types of antidiabetic medications have been developed and the management of patients with Type 2 diabetes mellitus (T2DM) has been substantially improved. While we can now successfully control hyperglycaemia in patients with T2DM, the number of patients with T2DM continues to rise. In addition, the financial cost of T2DM is a worldwide problem and cost-effective strategies for T2DM prevention are eagerly awaited. To develop and establish more effective prevention strategies for T2DM, this paper proposes a paradigm shift from a glucose-centric to a beta cell-centric concept of T2DM management. This concept makes it easier for medical staff and patients to understand the process of the development of T2DM and its complications in a pathophysiology-based, continuous, and integrated manner; the glucose-centric concept has so far failed to emphasise the importance of intensive intervention before the onset of T2DM. It is hoped that this paradigm shift in the management of T2DM will foster the development of novel preventive strategies to effectively control this pandemic disease.
INTRODUCTION
Diabetes is defined as a chronic hyperglycaemic state due to insufficient insulin action. Although defective insulin secretion from pancreatic beta cells and reduced insulin action in the liver, skeletal muscle, and adipose tissue are well established mechanisms in the pathogenesis of Type 2 diabetes mellitus (T2DM), recent studies have suggested that other organs and tissues, such as the brain, gastrointestinal tract, pancreatic alpha cells, kidney, and vascular endothelial cells, are also involved in the pathogenesis of hyperglycaemia.1
Exploring the different mechanisms that cause hyperglycaemia has resulted in an increase in the number of therapeutic targets and the development of different types of novel glucose-lowering drugs. Two recently developed agents, sodium/glucose cotransporter 2 inhibitors and glucagon-like peptide-1 receptor agonists, have been shown to improve cardiovascular and renal outcomes in patients with T2DM,27 beginning a new era in the treatment of T2DM.
Nonetheless, the number of patients with T2DM continues to rise across the world, resulting in a huge economic burden.8 While we are succeeding in improving glycaemic control in patients with T2DM, it remains difficult to achieve cure or remission, suggesting the importance of T2DM prevention and that our understanding of the pathogenesis of this disease remains unsatisfactory. This paper proposes a paradigm shift in the concept of T2DM based on its pathogenesis, from glucose-centric to a novel, beta cell-centric concept, in order to establish more effective prevention strategies.
THE GLUCOSE-CENTRIC CONCEPT OF DIABETES
Currently, the diagnosis of diabetes is based on plasma glucose levels9,10 and the diagnostic threshold of plasma glucose has been set based on the incidence of retinopathy, one of the microvascular complications of diabetes.11,12 People with impaired fasting glycaemia (IFG) and/or impaired glucose tolerance (IGT), known as prediabetes, are thereby defined as a population with a high risk of developing T2DM, but with a less severe glucose intolerance compared to T2DM patients.9,10
As a result, using this glucose-centric concept of T2DM, patients with IFG or IGT are recognised as having a less severe condition than those with T2DM, often resulting in a delay in intensive intervention for this population. However, while the diagnostic criteria of diabetes are based on the risk of developing microvascular complications, individuals with IFG or IGT have been shown to be at a high risk of developing atherosclerotic cardiovascular disease (ASCVD).13,14 Recent studies have revealed that the effect of strict glycaemic control on cardiovascular outcomes is modest,15-18 although such control does effectively reduce the risk of developing microvascular complications.19,20 Thus, to improve cardiovascular outcomes and extend healthy longevity, more intensive intervention in individuals with IFG or IGT, who are at a high risk of ASCVD, is needed.
A Need for Multifactorial Intervention
Recent studies have shown that not only glycaemic control but also multifactorial interventions, including blood pressure and lipid control and smoking cessation, are important and effective for the improvement of cardiovascular outcomes and mortality,21-23 even though some of the interventions, including smoking cessation and the use of statins, diuretics, and beta-blockers, may worsen glucose tolerance.24-26 Given the current challenge of reducing ASCVD rates in patients with T2DM, a glucose-centric concept of diabetes may not appropriately capture its pathophysiology or lead to effective treatment strategies.
Patients with metabolic syndrome (MetS), which consists of visceral obesity, glucose intolerance, hypertension, and dyslipidaemia, have also been shown to have an increased risk of developing ASCVD, indicating that an overlap of multiple risk factors has a major impact on the progression of atherosclerosis.27 Therefore, intensive intervention to prevent the future development of ASCVD, as well as T2DM, is needed in patients with MetS who are also at a high risk of T2DM; however, since prediabetes is recognised as a milder form of T2DM in the glucose-centric concept, intensive treatment for these patients is often not considered.
What Are We Missing?
Although the importance of cardiovascular risk reduction beyond glycaemic control in patients with T2DM has been highlighted, it may be difficult to establish effective strategies to improve cardiovascular outcomes with a glucose-centric concept of T2DM. One significant difficulty is that classification of glucose intolerance based on the current threshold of plasma glucose level may become an obstacle in capturing the continuous pathophysiology underlying the development of T2DM and its complications. To facilitate a more effective approach for achieving healthy longevity, which is the goal of diabetes care, a more continuous, comprehensive, and integrated concept of T2DM is needed.
BETA CELLS: THE CORE PATHOLOGY OF DIABETES
A number of organs and tissues regulate glucose metabolism in the body;1 however, it remains unclear as to what extent each mechanism is involved in physiological and pathological conditions. Recent genome-wide association studies have revealed a number of susceptible genes for T2DM, most of which are thought to associate with beta cell function or mass,28,29 indicating the major impact of beta cells on the development of T2DM.
In a physiological condition, when insulin sensitivity is reduced due to various factors, such as overnutrition, an inactive lifestyle, or obesity, insulin secretion increases to compensate and maintain normal glucose tolerance, resulting in a hyperinsulinaemic state. Hyperinsulinaemia is also often observed in patients with T2DM; however, studies have shown that disposition index, the true beta cell function adjusted for insulin sensitivity, is always reduced in patients with IFG, IGT, or T2DM,30,31 indicating that T2DM will not develop unless the compensatory mechanism of beta cells is impaired. In addition, histological studies have shown a reduction in beta cell mass in patients with IFG, IGT, or T2DM, irrespective of the presence of obesity.32,33 These findings highlight that a deficit of beta cell mass and/or function is common and the most important pathological feature of diabetes. Thus, this paper proposes a shift to a novel beta cell-centric concept of diabetes.
A PARADIGM SHIFT TO A BETA CELL-CENTRIC CONCEPT
Although T2DM is a progressive disease,34 this aspect makes the condition difficult to understand when using the categorical classification of diabetes based on plasma glucose level. Glycaemic control deteriorates with disease duration, which necessitates intensification of treatment; however, this nature of T2DM is related to progressive deterioration of beta cell function,35-37 which is thought to occur before and after the onset of T2DM.
Various mechanisms that induce beta cell dysfunction have been postulated, including gluco(lipo)toxicity,38 endoplasmic reticulum stress,39,40 oxidative stress,41 mitochondrial dysfunction,42 autophagy dysfunction,43 amyloid toxicity,44,45 cytokine pathways,46 and beta cell dedifferentiation and transdifferentiation.47 Excess beta cell workload due to a compensatory increase in insulin secretion in response to reduced insulin sensitivity may induce beta cell dysfunction and/or death through any of the aforementioned mechanisms, even before the development of hyperglycaemia.
In humans, the increase in beta cell mass in obese individuals is only modest,48,49 suggesting that the compensatory increase in insulin secretion in response to obesity is accomplished by an increase in insulin secretion from individual beta cells, which results in increased workload of these individual cells. Excess workload eventually induces beta cell dysfunction and/or death, leading to the development of hyperglycaemia.
Once beta cell mass is reduced, the workload on residual beta cells is further increased, resulting in a vicious cycle of beta cell dysfunction that may explain the progressive nature of T2DM. Therefore, from the viewpoint of the beta cell-centric concept, treatment strategies for T2DM should be directed towards a reduction in workload and protection of residual beta cells. To reduce workload, intensive intervention for patients with obesity and insulin resistance is critical.
In the beta cell-centric concept, pathophysiological changes prior to the onset of T2DM can be captured as a continuous process (Figure 1). Before the onset of T2DM, the reduction in beta cell mass is minimal, and improvement of obesity and insulin resistance at this stage could potentially normalise glucose metabolism.30,50,51 On the other hand, during this phase, compensatory hyperinsulinaemia, together with hypertension, dyslipidaemia and dysregulated adipokines, and inflammation due to visceral obesity, promotes atherosclerosis.52 Thus, timely intervention to improve obesity and insulin resistance through lifestyle modification is important because of two aspects: a) amelioration of hyperinsulinaemia will suppress progression of atherosclerosis, and b) reduction in beta cell workload will prevent beta cell loss and the development of T2DM. In this regard, assessment of insulin resistance in the non-diabetic population is important to identify individuals at a high risk of T2DM development, although obesity itself is already an established risk for ASCVD and most obese individuals also develop metabolic disorders in later life.53,54
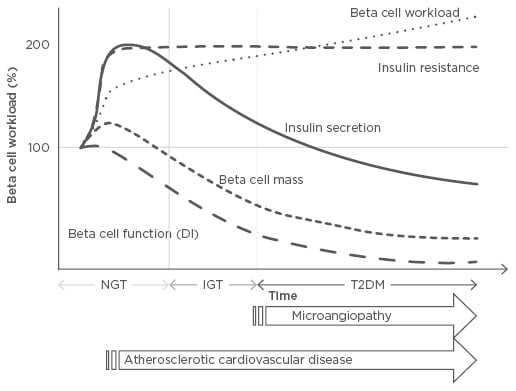
Figure 1: Changes in beta cell workload during the development of Type 2 diabetes mellitus.
Demand for increased insulin secretion upon insulin resistance due to excess caloric intake, physical inactivity, and obesity, but with a modest change in beta cell mass, results in an increase in individual beta cell workload. In this compensatory phase, the resulting hyperinsulinaemia, together with high blood pressure and dyslipidaemia, promotes atherosclerosis. Excess beta cell workload eventually leads to beta cell dysfunction and/or death, resulting in reduced beta cell functional mass. Once beta cell mass is reduced, workload on residual beta cells is further exaggerated, creating a vicious cycle. Finally, when beta cells fail to compensate, hyperglycaemia develops. After the development of diabetes, microvascular complications occur; therefore, to prevent both micro and macrovascular complications, reducing beta cell workload at an earlier stage prior to the onset of diabetes is needed.
DI: disposition index; IGT: impaired glucose tolerance; NGT: normal glucose tolerance; T2DM: Type 2 diabetes mellitus.
Moreover, amelioration of hyperinsulinaemia may reduce the risk of developing cancer and dementia, which is increased in patients with T2DM.55-57 The beta cell-centric concept is, therefore, a core therapeutic strategy to extend healthy longevity in people with and without diabetes (Figure 2).
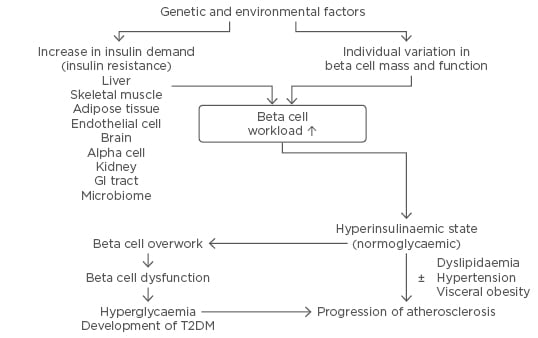
Figure 2: The beta cell-centric concept of Type 2 diabetes mellitus.
This concept emphasises the role of beta cells as the core pathoaetiological feature of T2DM and a therapeutic target to prevent the condition and its complications.
GI: gastrointestinal; T2DM: Type 2 diabetes mellitus.
CONCLUSION
A paradigm shift from a glucose-centric to a beta cell-centric concept of T2DM management will provide a continuous and integrated view of the pathophysiology involved in the development of T2DM. The beta cell-centric concept of diabetes has also recently been proposed and discussed in detail by other investigators.58-60 Continuous and integrated understanding of the process during the development of obesity, IFG or IGT, and T2DM, as well as the progression of microvascular complications and ASCVD, will lead to more vigorous interventions in clinical practice and efforts to explore novel therapeutic strategies aimed at beta cell protection. Sharing this concept with patients will also improve the decision-making process and adherence to treatment, including lifestyle modifications. To achieve this goal, comprehensive patient education is necessary.61
Various indices have been proposed to identify subjects at risk of T2DM, such as family history of T2DM, homeostasis model assessment indices, obesity and being overweight, MetS, HbA1c, fasting plasma glucose level, plasma glucose level at 2 hours during oral glucose tolerance test, insulinogenic index, and disposition index. However, further innovations, such as determining individual genetic risks for T2DM and ASCVD, identifying novel markers predicting beta cell workload within the normal range of plasma glucose levels using new technologies, such as metabolomics and proteomics, and developing a beta cell imaging technique to evaluate individual beta cell mass in vivo, will enable fostering of precision medicine and identification of high-risk individuals who need early intensive intervention.
In conclusion, obesity is an established risk factor for T2DM and ASCVD and is currently one of the largest healthcare problems across the world. Therefore, reducing the incidence of obesity through a healthy lifestyle is urgent. A paradigm shift from a glucose-centric to a beta cell-centric concept of T2DM will enable more comprehensive, integrative, and continuous intervention for individuals with obesity, IFG or IGT, and T2DM, and establish more effective prevention strategies for T2DM and its complications.