Abstract
Pituitary growth hormone (GH) is a peptide hormone predominantly secreted by somatotrophs in the anterior pituitary under the tight control of the hypothalamic–pituitary axis and GH secretagogues. GH elicits its effects directly on target organs and cells interacting with GH receptors and through stimulation of insulin-like growth factor 1 production. GH plays critical roles in regulating somatic growth and the metabolism of carbohydrates, lipids, and protein. GH increases insulin secretion and glucose uptake. Conversely, a GH deficient state is characterised by enhanced insulin sensitivity. Diabetogenic actions of GH are evident in conditions of GH excess, such as acromegaly or poorly controlled Type 1 diabetes mellitus. In patients with GH deficiency, administration of GH resulted in impaired glucose tolerance and insulin sensitivity. Owing to its multiple and complex effects, the regulation of GH secretion and its function in normal health and metabolic diseases is a major research interest in the field of molecular endocrinology. This review provides an overview of the effects of GH on glucose, lipid, and protein metabolism, insulin resistance, and metabolic homeostasis.
INTRODUCTION
Hormones control several steps of intermediary metabolism, including glucose oxidation, glycogen metabolism, gluconeogenesis, and fatty acid oxidation. The importance of hormones from the anterior pituitary, the islets of Langerhans, adrenal glands, and the thyroid in intermediary metabolism is well recognised. Over recent years there has been a significant increase in the understanding of how these hormones regulate metabolic homeostasis. An array of hormones, including insulin, glucagon, adrenaline, cortisol, thyroxin, amylin, glucagon-like peptide-1, glucose-dependent insulinotropic peptide, and pituitary growth hormone (GH), play prominent roles in the maintenance of glucose metabolism and homeostasis. Impaired glucose homeostasis is evident in several clinical conditions characterised by altered hormone levels, such as diabetes mellitus (Type 1 [T1DM] and Type 2), obesity, metabolic syndrome, hypothyroidism, Cushing’s disease, and acromegaly. Among the endocrine factors, pituitary GH deserves special attention owing to the myriad direct and indirect roles GH has on somatic growth, musculoskeletal anabolism,1 and regulation of carbohydrate, protein, lipid metabolism, and body fat distribution.2 The pituitary gland was first linked to carbohydrate metabolism when increased sensitivity to insulin was identified in hypophysectomised dogs, in a classic study by the Argentinian physiologist, Bernardo Alberto Houssay.3 Increased sensitivity to insulin was reversed when hypophysectomised animals were injected with pituitary extracts. A direct effect of GH was identified by demonstrating that injection of GH reduced glucose uptake in both adipose tissue and skeletal muscle.4 GH counteracts the overall effects of insulin on glucose and lipid metabolism;5 glucose uptake by skeletal muscle and adipocytes was blocked when both insulin and GH were co-administered.4
Acromegaly is a condition caused by elevated levels of circulating GH, usually secondary to a pituitary adenoma.6 In addition to inappropriate tissue and skeletal growth, acromegaly is characterised by perturbations of intermediary metabolism and diabetes.7 Ever since the Houssay studies showed reduced insulin requirement and improvement of hyperglycaemia in hypophysectomised animals,3 the influence of GH on glucose homeostasis has been explored by several studies.8-10 GH decreases glucose tolerance and glucose use that, in turn, results in compensatory hyperinsulinaemia. Prolonged administration of GH can induce diabetes in adult animals.11 A moderate elevation in the circulating levels of GH results in a spectrum of abnormal metabolic fuel concentrations associated with poor diabetes control. Patients with poorly controlled T1DM exhibit a 2–3-fold increase in GH levels that result in significant metabolic abnormalities.12 Thus, the hypersecretion of GH in T1DM may serve as a major contributor of poor glycaemic status associated with the disease.13 Humans with GH deficiency exhibit a higher BMI with obesity, hypertension, and elevated C-reactive protein levels. GH deficiency is associated with increased visceral adipose tissue.14 Treatment with either recombinant human GH (rhGH) or enhanced endogenous GH secretion affects glucose metabolism, reduces visceral fat, and ameliorates dyslipidaemia.15 This review discusses the metabolic perturbations such as hyperglycaemia, hyperinsulinaemia, and insulin resistance that arise due to alterations in GH levels, including the conditions of GH excess (acromegaly and T1DM) and GH deficiency.
GROWTH HORMONE SECRETION
GH secretion occurs episodically and is primarily under the control of two hypothalamic neuroendocrine hormones: GH-releasing hormone (GHRH), which stimulates GH secretion, and somatostatin, which inhibits GH secretion (Figure 1). Several other endocrine mediators also regulate GH gene transcription, including insulin-like growth factor 1 (IGF-1). IGF-1 is a major suppressor of GH transcription, whereas thyroxine, glucocorticoids, and ghrelin stimulate GH secretion. GH secretion also exhibits sexual dimorphism due to differential effects of oestrogen versus the androgens on GH. Pulsatile GH secretion occurs with a major surge at the onset of slow-wave sleep with less noticeable secretory episodes a few hours after meals. In healthy individuals, GH pulses discharge every 1–3 hours at an average of 45 μg per pulse, which equates to approximately 400–500 μg released every 24 hours, which occur as pulses within pulses. Autocrine secretion of GH by non-pituitary sources, such as mammary epithelial cells, have also been reported.16
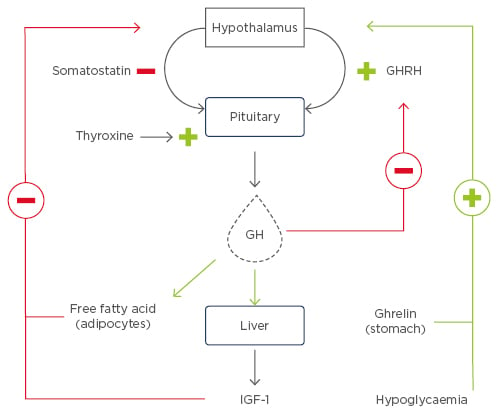
Figure 1: Factors regulating growth hormone secretion.
GH secretion by the pituitary gland is regulated by two hypothalamic hormones: GHRH, which has a stimulatory action at the level of gene transcription, and somatostatin, which has an inhibitory effect on GH secretion. IGF-1 plays a crucial role in regulating GH. IGF-1 is secreted by the liver under the influence of GH; however, IGF-1 also regulates GH release via a negative feedback loop. Elevated levels of circulatory IGF-1 stimulates somatostatin, thus resulting in decreased secretion of GH. High levels of GH inhibit GHRH. Ghrelin is a gastric hormone that positively regulates GH release, whereas plasma free fatty acids and low glucose levels also stimulate GH under physiological conditions.
GH: growth hormone; GHRH: growth hormone-releasing hormone; IGF-1: insulin-like growth factor.
In circulation, the preponderance of GH binds to the GH binding protein (GHBP), which regulates GH bioavailability. GHBP corresponds to the extracellular domain of the GH receptor (GHR) and is generated by proteolytic cleavage of the extracellular domain of the GHR in humans. In contrast, in rodents, alternative splicing of the GHR transcript generates GHBP. Hepatic IGF-1 secretion is one of the best-characterised effects of GH action. IGF-1 is also produced locally by tissues and elicits its effect via a paracrine and/or an autocrine manner. IGF-1 plays a critical role in inhibiting the secretion of the GH via negative feedback by stimulating somatostatin and inhibiting GHRH release (Figure 1). IGF-1 also has a direct effect on GH secretion, which is independent of GHRH and somatostatin. It is noteworthy that GH and IGF-1 secretion are regulated by each other, where the former induces the latter and the latter induces the former in a feedback loop. In healthy individuals, GH secretion is inhibited by hyperglycaemia and stimulated by sleep, stress, exercise, hypoglycaemia, and amino acids.
GROWTH HORMONE SIGNALLING
GH exerts its pleiotropic effects by binding to GHR. The prevailing model is that GHR exists as an inactive dimer and a single molecule of GH binds to two GHR molecules. The binding of the GH molecule to the extracellular domain of the GHR facilitates rotation of the two GHR molecules that results in the intracellular domain of each GHR molecules binding to a JAK2 molecule.17 JAK2 constitutively associates with the cytoplasmic domain of GHR and in the unliganded states the pseudokinase domain of JAK2 masks its catalytic domain. Binding of GH to the GHR re-orientates and rotates the receptor subunits, which results in the transition from parallel transmembrane domains to one where the transmembrane domains separate at the point of entry into the cytoplasm.18 This arrangement facilitates the movement of the pseudokinase inhibitory domain of one JAK2 away from the kinase domain of the other JAK2 within the receptor dimer–JAK2 complex. This results in the transactivation of JAK2. Activated JAK2 then phosphorylates the cytoplasmic domain of the GHR, which then recruits several downstream proteins as depicted in Figure 2.
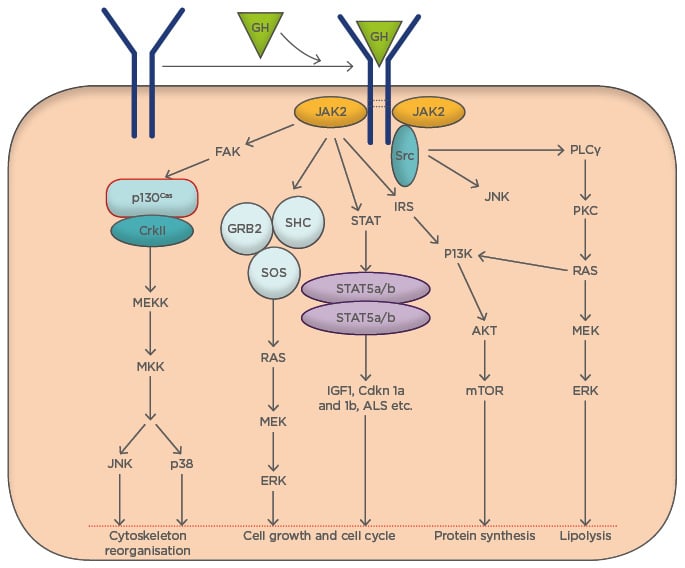
Figure 2: Cellular and molecular events triggered by growth hormone signalling.
GH acts via a variety of signal transduction pathways. Multiple GH signalling pathways can contribute to specific GH responses. GH binds to GHR and activates JAK2, which in turn triggers an array of signalling cascades. These interconnected signal transduction pathways regulate various metabolic and cellular events.
FAK: focal adhesion kinase; GH: growth hormone; GHR: growth hormone receptor; GRB2-SOS: growth factor receptor-bound 2-son of sevenless complex; IRS: insulin receptor substrate; PI3K: phosphatidylinositol 3-kinase; SH2-Bβ: src-homology 2 domain Bβ; STAT: signal transducer and activator of transcription.
Activation of JAK2 is a critical step for triggering GHR signalling. The STAT family of transcription factors is recruited to the activated GHR–JAK2 complex. STAT proteins are phosphorylated at a single tyrosine residue by JAK2. Phospho-STAT then undergo either homo or hetero-dimerisation and migrate to the nucleus where they act as transcription factors. Among various STAT proteins, STAT5b exerts the majority of the biological effects of GH. GHR can also activate MAPK, downstream of both JAK2 and Src kinase. GHR localisation to the lipid raft preferentially activates MAPK while cytosolic GHR localisation activates STAT5. Insulin receptor substrate-1 (IRS-1) plays a critical role in GHR-induced MAPK activation.19 Furthermore, GHR signalling is also associated with the activation of the phosphatidylinositol-3 kinase (PI3K)/Akt pathway in a JAK2/IRS-1-dependent manner. Attenuation of GHR activation is arbitrated by the proteins of suppressor of cytokine signalling (SOCS) family, which includes SOCS-1–7, and cytokine-inducible SH2-domain containing protein. It is noteworthy that SOCS family proteins are also induced by the JAK/STAT signalling cascade. GH itself has been shown to induce SOCS-2 and SOCS-3. SOCS proteins suppress GH signalling by inhibiting JAK2 activity and compete with STAT for binding with GHR or by inducing the proteasomal degradation of the GHR complex. Additionally, protein tyrosine phosphatases have also been implicated in terminating the GHR signal cascade.20 JAK2 binding stabilises and prevents GHR from degrading;21 on the other hand, GH induces desensitisation of the GHR via JAK2 kinase activity.22
GROWTH HORMONE AND CARBOHYDRATE METABOLISM
The pioneering work of Houssay established the role of anterior pituitary in the carbohydrate metabolism.3 The two classical findings of Houssay include hypophysectomy ameliorated the pancreatectomy-induced diabetes in dogs, and administration of the anterior pituitary extract induced hyperglycaemia, glycosuria, and ketonuria in normal dogs.3 Permanent diabetes was produced in dogs by injecting anterior pituitary extract.23 Histological studies in the dogs that received pituitary extract revealed degeneration of the islets of Langerhans.24 The diabetogenic effect of GH is further evidenced by studies that used purified GH. Several researchers, including Young25 and Campbell et al.,26 were able to induce diabetes in dogs and cats by administering purified GH.11 Furthermore, it was demonstrated that GH can induce diabetes in pancreatectomised animals.27 Hypophysectomy results in increased insulin sensitivity and prolonged GH administration abrogate the increase in insulin sensitivity.28 In hypophysectomised animals, administration of GH elicits glucose intolerance and insulin resistance. Diaphragms from hypophysectomised rats display decreased glucose uptake when treated with GH, compared with diaphragms from hypophysectomised rats naïve to GH treatment.29 GH inhibits the peripheral utilisation of sugars, thereby antagonising the action of insulin and serving to raise the blood glucose levels. Injection of GH into the brachial artery reduced forearm glucose uptake in both skeletal muscle and adipose tissue and blocked the action of insulin when both hormones were co-administered.4
The major steps in glucose metabolism include glucose uptake by tissues, such as the liver, muscles, and adipose tissue; glucose oxidation; glycogenesis; glycogenolysis; and gluconeogenesis. The precise role of GH on glucose uptake by tissues that play key roles in fuel homeostasis is unclear. Increased hepatic glucose uptake and glycogenesis were noticed in transgenic rats overexpressing the hGH gene, while insulin-dependent glucose uptake by adipocytes and muscle was impaired.30 In hGH transgenic mice, glucose infusion rate increased to 4.4 mg/kg/min, compared to 3.1 mg/kg/min in wild-type mice, and endogenous glucose production was suppressed with pegvisomant (an GHR antagonist) treatment in acromegalic patients assessed by the euglycemic clamp technique.31 Therefore, it was argued that the role of GH was to increase endogenous glucose production while its role in glucose utilisation is limited. Short-term exposure to GH inhibited insulin-stimulated glucose disposal by 27% and also blunted glycogen synthase activity in skeletal muscle.32
It has been established that GH stimulates glucose production in the liver, but it is unclear whether GH preferentially stimulates glycogenolysis or gluconeogenesis. Administration of rhGH to HIV patients for 6 months increased gluconeogenesis but not glycogenolysis.33 In healthy volunteers, GH administration (40 ng/kg/minute for 4 hours) increased glycogenolysis without altering the rate of gluconeogenesis.34 In a cohort of patients with acromegaly studied pre and post-pituitary microsurgery, glycogenolysis decreased by 29%, whereas gluconeogenesis remained unchanged, suggesting that GH preferentially increases glycogenolysis.35 It is noteworthy that the expression of gluconeogenic genes was not altered in GHR-/- mice compared with wild-type mice.36 These observations present a case for the fact that GH has a preferential effect on glycogenolysis compared to gluconeogenesis.
Contrary to the aforementioned studies, short-term administration of rhGH in lactating women increased gluconeogenesis, but not glycogenolysis.37 GH induces gluconeogenesis and glucose cycling in several organs, including splanchnic tissues, the liver, and adipose tissue. In healthy men, overnight administration of GH stimulates gluconeogenesis. Dogs treated with high doses (1 mg/kg/day) of GH for several days had increased liver glycogen content.38 Although baseline uptake of glucose by muscle tissue is low, a further suppression of glucose uptake is typically seen immediately following GH administration. Concomitant with decreased glucose oxidation, suppressed muscle glucose uptake in the presence of unchanged glucose turnover strongly suggests that GH promotes non-oxidative glucose disposal in a non-muscle compartment of the body.39 It is well established that intact GH secretion is crucial to combat prolonged hypoglycaemia, and GH is implicated as being a major mediator of the nocturnal increase in insulin requirements (also known as the dawn phenomenon) in patients with insulin-dependent diabetes.
GROWTH HORMONE AND INSULIN SENSITIVITY
In general, GH is thought to increase insulin resistance. Insulin resistance during puberty and pregnancy is, in part, attributed to increased concentrations of circulating GH. Insulin resistance is one of the clinical manifestations of GH treatment in humans. Administration of GH either within physiological range or supraphysiological doses leads to both hepatic and peripheral insulin resistance in healthy volunteers.40,41 The tissue that contributed most to the decreased insulin sensitivity was the muscle. Evidence for the direct effect of GH on insulin sensitivity has also come from studies in acromegaly subjects. Blocking GHR activation by pegvisomant for 4 weeks improved peripheral and hepatic insulin sensitivity in acromegalics.31 Although a GH-induced increase in free fatty acids (FFA) flux from the adipose tissue is associated with impaired insulin action at target tissues,42 studies in liver-specific IGF-1 deficient mice revealed a 3–4-fold increase in circulating GH levels and insulin resistance, without a significant increase in circulating FFA.43 Therefore, in addition to the increase in FFA, other factors also contribute to insulin resistance in GH elevated conditions. The possible mechanisms of GH-induced insulin resistance are presented in Figure 3, including SOCS-1 and 3-mediated insulin resistance and deregulation of insulin signalling.44 Interestingly, SOCS-2 serves as a negative regulator of GH signalling.45
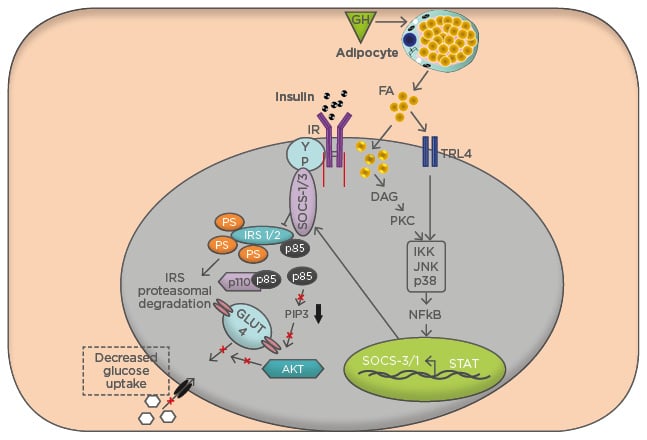
Figure 3: Mechanism of insulin resistance by excess growth hormone conditions.
The pathway for insulin stimulation of glucose transport in muscle involves activation of the insulin receptor protein, which docks IRS-1 and IRS-2 and phosphorylates these proteins on tyrosine residues (pY). IRS-1 recruits the p85α regulatory subunit of PI3K (p85-p110), resulting in phosphorylation of membrane-bound PIP3. Production of PIP3 is required for activation of Akt and signalling for GLUT4 translocation. Excess p85α acts as a dominant-negative signalling molecule by blocking the association of PI3K (p85-p110) with IRS-1 and thereby attenuating PI3K activation. Loss of PI3K activation from increased p85 and increased serine phosphorylation of IRS-1 both lead to reduced translocation of GLUT4 to the plasma membrane and result in decreased insulin-stimulated glucose uptake to skeletal muscle.
FA: fatty acid; GH: growth hormone; IR: insulin receptor; IRS: insulin receptor substrate; PIP3: phospholipids at the 3′ position; TLR: toll-like receptor.
GH elicits insulin resistance by increasing the expression of the p85α regulatory subunit of the PI3K.42 PI3K exists as a dimer of two subunits: p85 and p110; p85 is the regulatory subunit whereas p110 is the catalytic subunit. The p85 subunit binds to the insulin signalling molecule IRS-1 and inhibits insulin signalling. Heterozygous knockout of p85α improved insulin sensitivity in mice and complete absence of the p85 isoform resulted in hypoglycaemia.46,47 Hence, GH-induced expression of p85 would result in p85α homodimers competitively binding IRS-1 and blocking insulin signalling. Conversely, in GH deficient states, the p110 subunit binds to other p85 isoforms, such as p85β, and enhances insulin signalling. Transgenic mice expressing bovine GH have increased adipose tissue expression of a p85α subunit, concomitant with insulin resistance.42 Increased p85α in skeletal muscle induces insulin resistance in conditions of GH excess.48 Expression of p85 monomers is increased in transgenic mice overexpressing placental GH,49 and mice with a heterozygous deletion for p85α were protected from GH-induced insulin resistance.48 These multiple studies suggest that increased expression of p85α plays a critical role in GH-induced insulin resistance.
An alternative mechanism for GH-induced insulin resistance is via GH-dependent increase in plasma FFA. The inhibition of glucose oxidation by fatty acids is also termed the Randle cycle effect. Formation of TG from fatty acids results in accumulation of diacylglycerol and ceramides. These re-esterification intermediates activate protein kinase C isoforms, which downregulates insulin signalling by multiple mechanisms. GH replacement therapy is associated with inhibition of glucose metabolism as a result of the lipolytic effect of GH.50
A recent study conducted by the authors broadened their understanding of the biological roles of GH on insulin resistance in diet-induced obesity in mice. Under high-fat diet conditions, a lack of GHR in macrophages resulted in increased inflammation and decreased insulin sensitivity in adipose tissue.51 One of the physiological roles of GH in the macrophages is to attenuate NFκB activation and, in turn, suppress osteopontin expression. In the absence of functional GHR in macrophages, increased expression of osteopontin was associated with migration of inflammatory macrophages into adipose tissue and decreased insulin sensitivity in adipose tissue.51
GROWTH HORMONE AND LIPID METABOLISM
Acromegaly is characterised by profound disturbances of not only carbohydrate metabolism but also lipid metabolism. The principal effect of GH on lipid metabolism is to stimulate lipolysis and lipid oxidation, thereby sparing carbohydrates and proteins from immediate oxidative demands and constitutes a homeostatic mechanism.9 In addition to hyperinsulinaemia, patients with acromegaly have increased levels of circulating lipid intermediates, increased rates of lipid oxidation, and decreased rates of glucose oxidation.52 These lipid intermediates are absorbed by the muscle and interfere with glucose uptake and suppress glucose oxidation. To evaluate the importance of the raised GH levels in patients with poorly controlled diabetes, Press et al.13 injected GH at a rate of 100 μg pulses per hour for 45 hours into diabetic subjects. Plasma glucose concentrations doubled (86±11 to 204±17 mg/dL) within 8–10 hours and remained elevated until GH was discontinued. The hyperglycaemia was due to stimulation of hepatic glucose production in the absence of a significant change in levels of either insulin or glucagon. With GH administration, levels of circulating FFA, ketones, and branched-chain amino acids were also increased.13 This study suggested that in addition to hepatic glucose production, GH excess triggers ketosis and resulting acidosis. In another study, it was shown that administration of GH (210 µg) to diabetic subjects caused a significant increase in FFA (70%) levels and 3-hydroxybutyrate (400%) compared with subjects naïve to GH administration.52 Furthermore, increased non-oxidative glucose utilisation was observed and was evidenced by an increase in plasma lactate concentrations. GH-injected diabetic subjects also responded with increased lipid oxidation. These results suggest that GH is an important regulator of fuel fluxes in T1DM subjects, the key outcome being a transient stimulation of lipolysis.52 A steep increase in plasma FFA levels following a single GH pulse reflects the stimulation of lipolysis. Both pulsatile and continuous administration of moderate amounts of GH to healthy human volunteers lead to a dose-dependent stimulation of lipolysis as revealed by increased circulating FFA levels, glycerol, and increased lipid oxidation rates. The association of GH pulses with decreased rates of peripheral glucose uptake and increase in endogenous glucose production were entirely reversed by acipimox, an antilypolytic drug.53 A study in healthy adults showed that the nocturnal mean peak of GH precedes the increase in FFA by 2 hours,54 a time lag that mirrors the one observed after GH bolus administration,55 suggesting that GH is a regulator of diurnal fluctuations in oxidation rates of lipids and other fuel substrates.
Adiponectin, acting via the adiponectin receptor, increases fatty acid oxidation. GH increases expression of adiponectin receptor 2 in the liver.56 Exogenous GH administration to obese rats prevented hepatic steatosis and was associated with increased adiponectin receptor 2 expression.57 In addition to increased adiponectin receptor expression, decreased expression of peroxisome proliferator-activated receptors (PPAR)-α/β/γ isoforms in states of GH excess may facilitate enhanced triglycerides secretion. In PPAR-α knockout mice, a higher triglycerides secretion was observed with GH treatment than control mice. Furthermore, GHR knockout mice show decreased hepatic PPAR isoforms.58 GH stimulates lipolysis in visceral and subcutaneous adipose tissue by increasing the hormone-sensitive lipase activity, possibly by activating the β-adrenergic receptor. Similarly, there is evidence that suggests GH induces hormone-sensitive lipase in skeletal muscle.
GROWTH HORMONES AND PROTEIN METABOLISM
GH is an anabolic hormone that promotes protein synthesis both by decreasing the oxidation of amino acids and increasing the uptake of branched-chain amino acids by skeletal muscle.59,60 Under physiological states, the effect of GH on protein metabolism is limited to the augmentation of protein synthesis with a decreased breakdown at the whole-body level. GH mediated nitrogen retention is evidenced by reduced urinary excretion of urea, ammonium, and creatinine. GH induces protein synthesis by stimulating the mTOR/S6 kinase signalling pathway. Fasting is a state of GH resistance due to downregulation of GHR in the liver and other relevant tissues. In a fasting state, the decrease in GH action results in an increase in both protein catabolism by 25% and excretion of urea-nitrogen by 50%.61 Administration of GH to hypophysectomised rats restored the growth defects and decreased hepatic urea formation.62 GH administration contributed to muscle protein mass with no significant change in protein catabolism in malnourished haemodialysis patients.63 Age-related loss of muscle mass, also known as sarcopenia, correlates with diminished production of GH from the pituitary gland. Administration of GH induced accumulation of muscle mass during the aging process. Older rats who were administered GH exhibited improved sarcopenia associated with increased mitochondrial biogenesis and protection against oxidative damage in skeletal muscle by the induction of antioxidant enzymes.64
SUMMARY
To summarise, GH plays a complex role in the regulation of carbohydrate, lipid, and protein metabolism; increases circulating levels of glucose via increased endogenous glucose production; stimulates lipolysis to increase FFA, which in turn inhibits glucose oxidation; and impairs both hepatic and peripheral insulin sensitivity, particularly in the muscle where glucose oxidation is reduced. Therefore, GH reduces both insulin-dependent and glucose-dependent glucose disposal. The anabolic effect of GH on proteins is a consequence of the activation of lipolysis and the protein sparing actions of lipid fuels. To generalise, GH effects metabolism, it suppresses glucose uptake and glucose oxidation, and stimulates gluconeogenesis, glycogenesis, and lipolysis. GH antagonises the action of insulin on peripheral tissues and thereby decreases glucose uptake and increases glucose production. Insulin levels increase to counterbalance the elevated glucose following GH administration. GH-induced lipolysis in the visceral tissues and subsequent increased FFA interferes with insulin signalling pathways.
Although GH therapy in GH deficient adults reduces visceral adiposity, improves dyslipidaemia, and metabolic disturbances, GH treatment elicits impaired fasting glucose and increased insulin resistance, particularly in obese patients.65 Because insulin resistance and glucose intolerance are linked to the risk of diabetes, there is due concern that patients with metabolic disorders may develop diabetes following GH treatment. While short-term treatment with GH elicits insulin resistance in children and adolescents, the long-term consequences of GH replacement therapy on glucose metabolism is a matter of debate. Therefore, longitudinal studies with increased sample size are warranted to delineate the effect of GH administration on metabolic disorder and cardiovascular outcomes.