Abstract
Diabetes has become an emerging public health problem because of its serious complications, and high mortality and morbidity rates. Among the most common microvascular complications of diabetes is diabetic nephropathy (DN), which is a major cause of development of end-stage renal disease worldwide. The aetiopathogenesis of DN is not completely elucidated; however, studies have shown that the components of the MAPK signalling pathway play an essential role in the development and progression of the disease. The MAPK family is mainly composed of three subgroups: extracellular signal-regulated kinases 1 and 2, c-Jun N-terminal kinases (JNK) 1–3, and p38 MAPK, all of which are related to several cellular functions, such as cell death, differentiation, proliferation, motility, survival, stress response, and cell growth. In diabetic kidney disease, the MAPK pathway can be activated by processes resulting from hyperglycaemia (polyol pathway products, oxidative stress, and accumulation of advanced glycosylation end-products) and by angiotensin II, and it is related to several renal pathological processes. This review aims to summarise the role of the MAPK signalling pathway in diabetic nephropathy, as well as to link the biological aspects that contribute to clarify the pathological process behind the disease.
INTRODUCTION
Diabetic nephropathy (DN) is considered the most common microvascular complication of diabetes, being a major cause of end-stage renal disease (ESRD) and cardiovascular mortality.1-3 Data from the United States Renal Data System demonstrate that prevalence of cardiovascular disease is greater in patients with chronic kidney disease, which may present complications, such as acute myocardial infarction and atrial fibrillation.4 Patients with ESRD require dialysis or kidney transplant;1 thus, complications could lead to limitations in quality of life, both by the severity of the disease and by the number of hospitalisations.4 DN contributes to high costs in public healthcare, especially when it progresses to ESRD. Patients with DN have 50% higher annual costs than patients who present with diabetes alone, and patients with ESRD who undergo dialysis have 2.8-times the annual mean costs compared to those with ESRD and are not receiving dialytic treatment.5 Due to the severity of complications and socioeconomic factors related to the disease, many mechanisms are being studied to understand the aetiopathogenesis of the DN because it is still not fully elucidated.6 Factors such as genetic susceptibility, increased products of the polyol pathway, activation of the renin–angiotensin system, and increased production of advanced glycation end products (AGE) have been related to the physiopathology of the disease.6
It has also been demonstrated that the MAPK signalling pathway plays a crucial role in DN; therefore, it is important to investigate its relationship with the pathogenesis of the disease since this understanding can clarify the pathological process behind the disease and contribute to the development of new therapeutic strategies.7
MAPK SIGNALLING PATHWAY AND ITS BIOLOGICAL FUNCTIONS
MAPK are a family of serine/threonine protein kinases (Ser/Thr) responsible for promoting intracellular signal transduction from extracellular stimuli.8 In this way, they regulate innumerable and important cellular actions, such as cell death, differentiation, proliferation, motility, apoptosis, survival, and stress response.7,9 These proteins are activated by a series of cascade reactions and are composed of three core kinases that are subsequently phosphorylated: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK (Figure 1). This cascade has upstream and downstream elements, known as MAPK kinase kinase kinase (MAPKKKK) and MAPK activated protein kinase (MAPKAPK), respectively.9 The MAPK group is classified into three major subgroups: extracellular signal-regulated kinase (ERK) 1 and 2, c-Jun N-terminal kinases (JNK) 1–3, and p38 MAPK.7 ERK are activated by growth factors, while JNK and p38 MAPK are activated by cytokines, cell death receptors, mitogens, and stressors (oxidative stress, hypoxia, thermal shock, and ultraviolet radiation), and therefore also known as stress-activated protein kinases (SAPK) (Figure 1).10,11 ERK is an essential signalling pathway that controls cellular processes, such as proliferation, survival, and differentiation. Activation of ERK begins with the recruitment of the adapter protein growth factor receptor-bound protein 2 (Grb2) and Son of Sevenless (SOS), which propitiate the exchange of guanosine diphosphate (GDP) of the GTPase RAS protein to a GTP.12 Activated RAS stimulates the RAF (MAPKKK) by phosphorylation of two residues of Ser active MAPKK (MEK1/MEK2).9 Phosphorylated MEK1/MEK2 signals to ERK 1 and 2 (MAPK) via threonine and tyrosine residue phosphorylation (Thr and Tyr) in the threonine-glutamine-tyrosine domain (Thr-Glu-Tyr).9 The subgroups of MAPK act in the cellular nucleus in order to propitiate the activation of transcription factors (Figure 1).8,10 The different variants of the JNK pathway, 1, 2, and 3, are encoded by the JNK1, JNK2, and JNK3 genes, respectively, and are associated with inflammation, apoptosis, cell differentiation, and proliferation.11,13,14 After stimulation of this pathway, a series of events occur until the final formation of JNK.13 The initial step is the action of small GTPases, Cdc42, Rac, and Rho, in MAPKKK, which have 14 different forms capable of activating MAPKK (MKK4 and MKK7). These are MEKK1, MEKK2, MEKK4, MLK1, MLK2, MLK3, MLK4, DLK, LZK, ASK1, TAK1, TAO1, TAO2, and ZAK.13-15 After activation, MKK4 and MKK7, either separately or in combination, are capable of phosphorylation of the residues Thr and Tyr and all three isoforms of JNK (Figure 1).14 The p38 MAPK cascade presents a series of components shared with JNK, having similar biological functions, such as inflammation, differentiation, proliferation, and apoptosis (Figure 1).13 This is related to regulation of the production of proinflammatory cytokines, such as TNF-α and IL-1β, and is therefore of paramount importance for the control of immunological effects.16 It is of importance that the mammalian p38 pathway presents four different isoforms: α, β, γ, and δ, which are encoded by the MAPK14, MAPK11, MAPK12, and MAPK13 genes, respectively.17 The α and β isoforms are expressed in several tissues, including in the brain, whereas γ and δ are associated with specific tissues.16
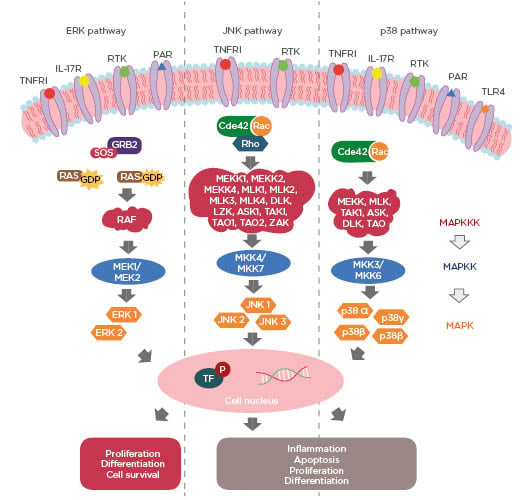
Figure 1: Three major pathways of MAPK: extracellular signal-regulated kinase (ERK) 1 and 2, c-Jun N-terminal kinases (JNK) 1-3 and p38 MAPK.
Cdc42: cell division control protein 42; GDP: guanosine diphosphate; GTP: guanosine triphosphate; GRB2: growth factor receptor-bound protein 2; ERK: extracellular-signal-regulated kinase; JNK: c-Jun N-terminal kinases; MAPK: MAP kinase; MAPKK: MAP kinase kinase; MAPKKK: MAPK kinase kinase kinase; P: phosphorylation; PAR: protease-activated receptor; RTK: tyrosine kinase receptors; SOS: Son of Sevenless; TF: transcription factors; TLR4: toll-like receptors 4; TNFR1: tumour necrosis factor receptor 1.
p38 MAPK can be activated by cytokines and stressor stimuli which act on small GTPases, Cdc42 and Rac, that are capable of activating one of the different forms of MAKKK (MEKK, MLK, TAK1, ASK, DLK, and TAO).11,13,15 These phosphorylate MAPKK (MKK 3/6) and transmit the signal to the different isoforms of p38 MAPK.10 Activation of all three major components of the MAPK pathway is complex and involves several receptors.10,18 Growth factors activate the MAPK ERK pathway via tyrosine kinase receptors (RTK), while inflammatory cytokines and stressors are activators of these receptors for p38 and JNK, respectively.18 MAPK function are activated downstream by TNFR1, IL-17 receptor, protease-activated receptor 1 (PAR1) and 4 (PAR4) and toll-like receptors 4 (TLR4) (Figure 1).10,19-22
DOES THE MAPK SIGNALLING PATHWAY INFLUENCE THE PATHOGENESIS OF DIABETIC NEPHROPATHY?
MAPK signalling pathway is related to inflammatory, oxidative, and apoptotic processes and therefore has a crucial role in DN development.8 Among mechanisms proposed to explain the activation of MAPK components in DN are insulin, high glucose levels, oxidative stress, inflammation, and angiotensin II.8,22-25 It is well known that diabetes promotes glucose cell deficiency, which is associated with insulin resistance and compensatory hyperinsulinaemia.26 Moreover, iatrogenic hyperinsulinaemia may occur in Type 1 and Type 2 diabetes mellitus by insulin treatment, one of the most effective strategies to delay the disease.23 The presence of insulin in both situations can lead to kidney damage through MAPK pathway activation.27
Insulin directly activates all three MAPK pathways and could also indirectly produce an excess of H2O2 in podocytes, a compound related to oxidative stress and therefore associated with activation of the MAPK pathway.23,24,27 Although ERK, JNK, and p38 MAPK are phosphorylated in response to insulin, the pathway that mediates this process is only well established for ERK1 and ERK2.27 This mechanism has a series of reactions that begins with the binding of insulin to its cognate receptor. The phosphorylation of the insulin receptor substrate (IRS) 1 and 2, which is an important adapter for Grb2 and SOS, results in ERK activation.27
Alongside hyperinsulinaemia, high glucose levels activate the MAPK cascade via many approaches, such as the polyol pathway, oxidative stress, and accumulation of AGE.28 It has been demonstrated that renal tubular epithelial cells (NRK-52E) exposed to a high glucose medium activated ERK, JNK, and p38 MAPK and that renal proximal tubular epithelial cells (HK‑2) incubated in high glucose concentration caused activation of the p38 MAPK pathway, which was associated with increased cell apoptosis.7,29 Activation of MAPK components is also mediated by inflammation, and vice versa.8,30 Inflammatory cytokines and growth factors, such as IL-1 and TNF-α are associated with interstitial fibrosis in diabetic patients and experimental models.21 TNF-α activates all three groups of MAPK signalling pathways, JNK and p38-MAPK are also stimulated by IL-1.10,30,31 Studies have shown that p38 MAPK and ERK are activated in human podocyte cells by IL-17RA and that p38 MAPK is phosphorylated in the kidney of diabetic rats and in rat mesangial cells (HBZY-1) by TLR4.19,22
Additional studies have revealed that angiotensin II activates ERK1 and ERK2 from proximal tubule cells in vitro through processes that include angiotensin II receptors.25,32,33 A model performed in the human embryonic kidney (293 cells) identified the fundamental role of angiotensin II and its receptor angiotensin Type II 1a receptor (AT1aR) in ERK1 and ERK2 activation. Stimulation of the receptor AT1aR by angiotensin II mediates downstream ERK phosphorylation by two distinct mechanisms: heterotrimeric guanine nucleotide-binding protein (G protein) and β-arrestin. The G protein (Gαq/11) stimulates protein kinase C (PKC) and downstream signalling that promotes activation of RAF-1, MEK, and ERK.34
Many other receptors with different functions are related to phosphorylation of the MAPK pathway in DN.19-21,35 In an in vitro study with mesangial cells, ERK 1 and ERK2 activation was PAR-1 dependent, related to fibronectin production by increasing TGF-β signalling.20 It has also been shown that the production of AGE stimulated the synthesis of kallikrein and activated PAR-4, promoting stimulation of the p38 MAPK pathway, which led to C-C Motif Chemokine Ligand 2 (CCL2), IL-8, and IL-6 production.21
The interaction of AGE with its cognate receptor (RAGE) promoted the production of reactive oxygen species in mesangial cells, which in turn stimulated the non-receptor Tyr kinases (NRTK) of the Src family. Consequently, Src/NRTK phosphorylated phosphatidylinositol 3-kinase (PI3K) to activate RAS, RAF, MEK, ERK1, and ERK2, leading the expression of insulin-like growth factor 1 (IGF-1), fibronectin, collagen, and TGF-β.35
MAPK phosphorylation is an event that promotes a series of reactions. The classical mechanism of action of MAPK is divided into two steps: firstly, its phosphorylation occurs in the cellular cytoplasm, and then it is translocated to the nucleus, where it activates several transcription factors through the phosphorylation of its Ser and Thr residues.8
Among the transcription factors stimulated by MAPK that mediate inflammation in DN is NF-κB.8 This protein is associated with increased extracellular matrix (ECM) accumulation and renal fibrosis through the production of TGF-β1;36 furthermore, NF-κB plays an important role in the production of inflammatory cytokines, such as TNF-α and IL-6, which are associated with the pathogenesis of DN.8
MAPK pathway inactivation contributes to reduced expression of inflammatory (TNF-α, NF-κB, and IL-6) and apoptotic elements (Bax and caspase-3) in DN, its phosphorylation is also related to the increase of adhesion molecules, which, in association with cytokines, promotes cell death.8
MAPK phosphorylation promotes apoptosis, infiltration of inflammatory cells, ECM synthesis, and renal inflammation and dysfunction, and is therefore important for kidney disease pathogenesis.7,8
A significant reduction of the fibrotic area of glomeruli and renal tubules occurred in a Type 1 diabetic mouse model through the use of JNK inhibitor SP600125, ERK inhibitor PD98059, and p38 inhibitor SB203850. Gene expression of TNF-α, IL-6, and iNOS was also decreased in renal tissues after treatment with these MAPK inhibitors.37 Though phosphorylated ERK and phosphorylated p38 were detected in various renal components of kidneys specimens of individuals with DN, such as podocytes, tubular epithelial cells, mesangial cells, and endothelial cells, only phosphorylated ERK correlated with the progression of glomerular lesions.38
In DN, ERK pathway elements (MEK 1, MEK 2, ERK 1 and ERK 2) were expressed more in the glomerular mesangial and epithelial renal cells of kidney tissues.39 In the context of hyperglycaemia, the JNK pathway is associated with mesangial cell proliferation and fibronectin expression, whereas p38 MAPK is predominantly expressed in diabetic tubules and therefore related to tubular lesions.29,40 TGF-β is located in this same p38 MAPK region, which indicates a correlation between p38 MAPK in TGF‑β‑mediated renal fibrosis.29
p38 MAPK induce renal tubular cell apoptosis and their suppression promotes a reduction in apoptosis and improves renal dysfunction.29 Phosphorylated p38 MAPK also increases levels of TNF-α, IL-1, IL-6, and IL-8, promoting glomerulonephritis and DN development;41 thus, inhibition of the p38 MAPK pathway promotes the reduction of inflammation and fibrosis, improving manifestations of kidney disease.30,37
The MAPK pathway directly promotes the renin–angiotensin–aldosterone system (RAAS), which contributes to the pathogenesis of DN.7,37 ERK1, ERK2, JNK, and the p38 MAPK pathway were directly involved in angiotensin-converting enzyme (ACE) production in a study in vitro with renal tubular epithelial cells (NRK-52E).7 Similarly, administration of MAPK inhibitors promoted reduced gene expression of angiotensinogen or renin in renal tissues, and JNK signalling was especially associated with downregulation of the ACE/angiotensin II pathway.37
Several studies about possible therapeutic strategies for inhibition of MAPK cascade in DN have been developed in recent years (Table 1).7,8,42,43 It was demonstrated that two drugs, metformin and glucagon-like peptide-1–based therapies (GLP-1), already effective in treating diabetes, also corroborate the improvement of DN by inhibiting components of the MAPK pathway.44-47 A study in vitro showed that metformin inhibits ERK1, ERK2, and p38 MAPK phosphorylation in mouse mesangial cells exposed to high glucose levels, while a streptozotocin-induced diabetic nephropathy rat model provided evidence that GLP-1 inhibited p38 MAPK activity via GLP1-receptor, which improves inflammation and fibrosis in the kidneys.44-45
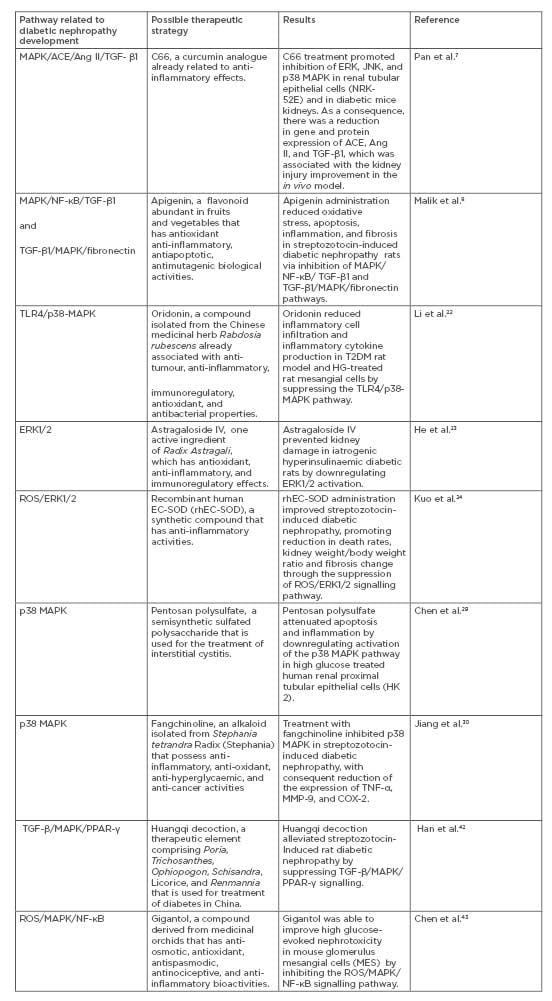
Table 1: Possible therapeutic strategies for inhibition of MAPK cascade in DN.
ACE: Angiotensin-converting enzyme; Ang II: Angiontesin II; C66: (2E,6E)-2,6-bis(2-(trifluoromethyl) benzylidene)cyclohexanone; DN: diabetic nephropathy; ERK: extracellular signal-regulated kinase; JNK: c-Jun N-terminal kinase; PPAR-γ: receptor peroxisome proliferator-activated receptor-γ; ROS: reactive oxygen species; TGF-β1: transforming growth factor beta 1; TLR4: toll-like receptors 4.
CONCLUSION
DN is a serious complication of diabetes and directly contributes to the development of chronic kidney disease. This is associated with high mortality, morbidity, and high costs in public healthcare; therefore, it is important to understand its aetiopathogenesis because this is not yet completely elucidated. It has been demonstrated that the pathogenesis of DN is complex and involves several interactions and mechanisms, such as the MAPK signalling pathway. The MAPK signalling pathway is crucial for cellular responses and in DN it can be affected by many factors that are closely associated with the pathophysiology of the disease. Stimulation of the pathway promotes deregulation of renal tissue function, which in turn promotes a pathological process that leads to kidney fibrosis, contributing to the development of ESRD. Understanding the association of this biological pathway with DN is fundamental for better clarification of the aetiopathogenesis of the disease and for the development of new therapeutic strategies for prevention, improved quality of life, and the reduction of costs for public health.