Abstract
Gestational diabetes mellitus (GDM) is a pregnancy complication defined as a glucose intolerance of varying severity with onset or first recognition during pregnancy. The prevalence of GDM is growing rapidly worldwide. Two major metabolic disorders, chronic insulin resistance and β-cell dysfunction, are currently linked to the pathogenesis of GDM, although the cellular mechanisms involved are not yet completely understood. Maternal genetic predisposition coupled with environmental and fetoplacental factors initiates a chain of events that affects mother and fetus, both in the short and long term. Understanding of pathophysiology and risk factors will enhance the possibility of effective screening, early intervention, and even prevention.
INTRODUCTION
Gestational diabetes mellitus (GDM) is defined as a glucose intolerance of varying severity with onset or first recognition during pregnancy. According to the 2017 International Diabetes Federation (IDF) estimates, GDM affects 14% of pregnancies worldwide, representing ~18.4 million births annually.1 GDM develops when insulin secretion fails to overcome the physiologic insulin resistance (IR) during pregnancy.2 Known risk factors, such as advanced maternal age, overweight, obesity, westernised diet, ethnicity, intrauterine environment, hypertension, family history of GDM or Type 2 diabetes mellitus (T2DM), and personal history of GDM or polycystic ovarian syndrome, are either directly or indirectly associated with impaired β-cell function and/or insulin sensitivity. Therefore, GDM may represent a transient ‘unmasking’ of pre-existing latent metabolic disturbances. Maternal genetic predisposition coupled with environmental and fetoplacental factors initiates a chain of events affecting mother and fetus in both the short and long term.2 Understanding of the pathophysiology will enhance the possibility of effective screening, early intervention, and even prevention. This review will discuss molecular processes underlying the pathophysiology of GDM. Potential mechanisms behind GDM are summarised in Table 1.
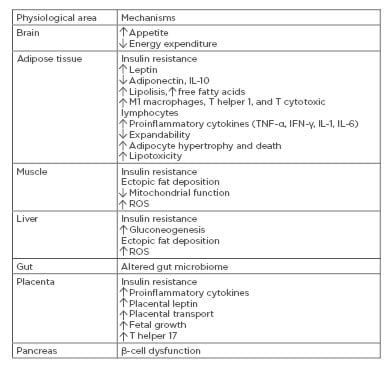
Table 1: Potential mechanisms behind gestational diabetes mellitus.
IFN-γ: interferon-γ; ROS: reactive oxygen species.
MATERNAL PREDISPOSITION OR PREGNANCY-MEDIATED EXACERBATION?
IR and β-cell dysfunction are critical pathophysiologic components of both GDM and T2DM. These impairments exist prior to pregnancy and can be progressive, representing an increased risk of T2DM after pregnancy,
ranging between 17% and 63% within 15 years.3 The development of both diseases is governed by a complex interaction of environmental factors with multiple genes. Polymorphisms of susceptible genes of T2DM have been shown to relate to development of GDM, suggesting a considerable overlap between the genetic contributors.4 The majority of susceptibility genes are related to β-cell function, including KCNJ11, KCNQ1, MTNR1B, IGF2BP2, and rs7754840 and rs7756992 in CDKAL1.4
During pregnancy, the combination of hormonal changes and disturbed endocrine function of adipose tissue and placenta are added to genetic predisposition and environmental factors (Figure 1). There is a uniform 50–60% decrease in insulin sensitivity with advancing gestation in both normoglycaemic and diabetic women.5 The decreased insulin sensitivity in GDM, however, occurs with a background of chronic IR preceding pregnancy to which the physiological IR of pregnancy is partially additive.5 Thus, the metabolic and endocrine changes accompanying the second half of gestation, inducing physiological IR, unmask and worsen the underlying pre-existing metabolic disturbances, leading to the full clinical picture of GDM.6 The physiological IR abates rapidly following delivery, but women with GDM end up, on average, with considerably greater IR than normal women.
The pathophysiological changes responsible for IR in GDM are not fully clarified. Insulin receptor abundance is usually unaffected.7 Genetic variants associated with IR and abnormal use of glucose (PPARG, TCF7L2, GCK, GCKR) show associations with GDM risk.4
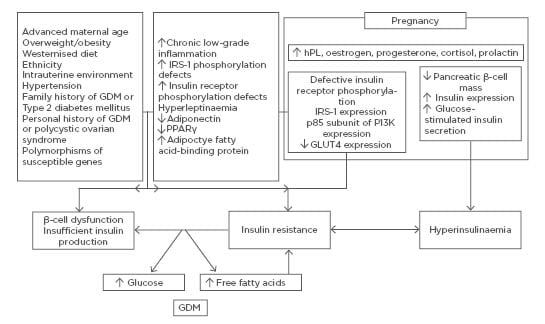
Figure 1: Mechanisms underlying insulin resistance in normal pregnancy and gestational diabetes mellitus.
A pronounced physiological decrease in peripheral insulin sensitivity occurs as pregnancy proceeds. This decrease is mediated by a number of factors such as increase in the levels of hPL, oestrogen, progesterone, cortisol, and prolactin, among other factors. In addition to altering components of peripheral insulin signalling cascades, they also activate various mechanisms enhancing β-cell function. GDM develops when insulin secretion fails to overcome the physiologic insulin resistance during pregnancy. The metabolic/endocrine changes accompanying the second half of gestation and inducing physiological insulin resistance unmask and worsen the underlying pre-existing metabolic disturbances, leading to the full clinical picture of GDM.
GDM: gestational diabetes mellitus; hPL: human placental lactogen; IRS-1: insulin receptor substrate 1; PPAR-γ: peroxisome proliferator-activated receptor gamma.
Altered expression and/or phosphorylation of downstream regulators of insulin signalling, including insulin receptor substrate (IRS)-1, phosphatidylinositol 3-kinase (PI3K), and glucose transporter (GLUT)-4, reduced expression of peroxisome proliferator-activated receptor (PPAR)-γ, and increased expression of the membrane glycoprotein PC-1 have been described during and beyond pregnancy.6,7 IR exacerbates β-cell dysfunction.2
PLACENTAL VERSUS MATERNAL TISSUES ROLE IN GESTATIONAL DIABETES MELLITUS
At the onset of gestation, the interaction of maternal insulin with the syncytiotrophoblast may lead to altered synthesis and secretion of cytokines that, in turn, will act on the mother, thus forming a feedback loop. This functional interplay contributes to the low-grade systemic inflammation during the third trimester of uncomplicated pregnancy by expressing a common repertoire of cytokines.8 This situation is exacerbated in obese pregnancies and those with GDM.8 However, the relative contribution of maternal adipose tissue, as well as the placenta, to this inflammation is yet to be determined.
ROLE OF FETOPLACENTAL UNIT IN GESTATIONAL DIABETES MELLITUS
Placental-Derived Exosomes
Placental-derived exosomes play a role in maternal immunomodulation through suppression of natural killer cell and macrophage activation, and induction of lymphocyte apoptosis.9 They suppress T-signalling components such as CD3-zeta and JAK3, while inducing suppressor of cytokine signalling (SOCS)-2.10 This counteracts allograft rejection of the fetus and sustains cellular adaptation in the face of physiological changes associated with pregnancy. Placenta-derived exosomes carry syncytin-1, which mediates trophoblastic syncytialisation and regulates endothelial cell migration, thereby sculpting the maternal-fetal circulation.11
The total number of exosomes in maternal plasma between 11 and 14 weeks of gestation is up to 2-fold greater in women with GDM.12 The released exosomes from trophoblasts in GDM subjects induce secretion of proinflammatory cytokines from endothelial cells.12 Furthermore, upregulation of microRNA-326 in diabetic patients in a comparison analysis negatively correlated with its target, adiponectin.13 This mechanism potentially mediates the inflammatory phenomena typically associated with GDM. However, with regard to several microRNA candidates as biomarkers for GDM, the results are often discordant. Exposure to bisphenol A has been associated with GDM and with alterations in methylation.14 This could be because bisphenol A induces exosome signalling from the placenta.
Hormonal Effect in Gestational Diabetes Mellitus
Progesterone is a major contributor to IR whereas oestrogen is protective.15 Since oestrogen receptor-α appears to play a major role in adipose tissue, its decreased expression in subcutaneous fat of patients with GDM may contribute to the development of IR.16 Additionally, progesterone may prohibit normal adaptation of β-cell reserves during pregnancy and affect the susceptibility to diabetes.17 An observational study found significantly higher levels of serum cortisol in women with GDM.18 In skeletal muscle, glucocorticoids induce IR by decreasing transcription of IRS-1, while increasing transcription of protein tyrosine phosphatase type-1B and p38 mitogen-activated protein kinase (MAPK) which counteract insulin action.19
Human placental lactogen (hPL) displays both insulin-like and anti-insulin effects. It acts via the prolactin receptor (PRLR) on maternal β-cells to mediate increases in β-cell mass and function, and to address the increased metabolic demands of pregnancy.20 In contrast, it causes profound IR by decreasing phosphorylation of IRS-1.20 Additionally, the hPL-related increase in free fatty acid (FA) levels directly interferes with insulin-directed entry of glucose into cells. Placental hPL messenger RNA levels were found to be higher in GDM patients;21 therefore, hPL is considered as a major, physiologic antagonist to insulin action during pregnancy.21
Human placental growth hormone (hPGH) may contribute to IR by specifically increasing the expression of the p85-regulatory unit of PI3K, resulting in a marked reduction in IRS-1-associated PI3K activity.22 In contrast, treatment with the 20-kDa hPGH-variant was suggestive of enhanced insulin sensitivity in an animal trial. It demonstrated diminished diabetogenic properties compared to the native pituitary 22-kDa hGH-N.23
Prolactin largely regulates its functions on β-cell via the JAK-2/signal transducer and activator of transcription (STAT)-5 pathway.24 Pathway disruption and single nucleotide polymorphisms of the 5′-untranslated region and promoter region of the PRLR gene have been shown to be associated with increased risk for GDM.25 On the contrary, another study found no difference in prolactin concentrations before and during the oral glucose tolerance tests between normal and diabetic pregnancies, neither in pregnancy nor postpartum.26 There was no correlation between the deterioration in glucose tolerance and the prolactin concentrations in either group. These discrepancies may be attributable to the complex crosstalk between PRLR and insulin receptors. Further research is required to determine the physiopathological importance of prolactin in the development of GDM.
Placental Inflammation and Adipokines in Gestational Diabetes Mellitus
The placenta may play an active role in mediating inflammation in women with obesity and GDM. Women with GDM in the third trimester of pregnancy had a higher proportion of T-helper 17 cells (proinflammatory response) which was balanced with a parallel higher proportion of T-helper 2 and regulatory T cells (anti-inflammatory response).27 The co-operation between different maternal and fetal cell types may propagate a vicious cycle for enhancement of cytokine production. This condition can alter placental metabolic and endocrine functions, which are likely to contribute to the placental changes, and eventually affect insulin action at the fetoplacental unit leading to GDM in utero.28 Among pathways upregulated by GDM insult in placentas, TNF-α signalling is one of the most relevant to placental growth;29 additionally, it links inflammation to defective insulin action and may reinforce the endocrine mechanism of pregnancy-induced IR by adding a placental component.28 Elevated TNF-α transcription in the placenta has been associated with markedly decreased insulin signalling in GDM.28 It is possible that TNF-α downregulates insulin action through serine phosphorylation of placental insulin receptors.28
Hyperleptinaemia may also contribute to enhanced IR in the GDM mother.30 Increased placental leptin expression and synthesis in GDM amplifies low-grade inflammation by stimulating production of both CC-chemokine ligands and proinflammatory cytokines, which further enhances leptin production;30 therefore, a vicious circle develops, aggravating the inflammation. However, it would be challenging to differentiate the relative contribution of placental versus maternal tissues for regulation through TNF-α and leptin. The effect of adiponectin that may be implicated in IR with advancing gestation in normal and GDM pregnancies through a decrease in maternal concentrations is exclusively of maternal origin due to the absence of ligand, but expression of adiponectin receptors in the placenta.30
Proinflammatory cytokines upregulate insulin-stimulated amino acid transporter system-A activity, while IL-1β downregulates it in cultured primary human trophoblasts.31,32 Additionally, leptin and TNF-α activate phospholipase A2, which generates docosahexaenoic acid, an essential ω-3 polyunsaturated FA. An accumulation of ω-3 FA has been recently demonstrated in the placenta of offspring of GDM mothers with increased adiposity at birth.33 These may be potential mechanisms by which local placental inflammatory mediators and responses influence placental nutrient transport and increase lipid substrate availability for fetal fat deposition, thereby linking inflammation in maternal obesity to changes in fetal growth. However, maternal inflammation in obese women or women with GDM may influence fetal development by impacting placental function, rather than directly influencing the fetal inflammatory profile.33 Most maternal cytokines do not cross the trophoblast barrier and, hence, do not reach the fetal circulation.34 It is therefore possible that the placenta acts as a mediator and an adaptor in pregnancy, sensing and responding to the maternal inflammatory environment to maintain pregnancy and to limit exposure of the fetus to inflammation and oxidative stress despite GDM insult.34
ROLE OF MATERNAL ADIPOSE TISSUE IN GESTATIONAL DIABETES MELLITUS
Neurohormonal Network
The neurohormonal network regulates appetite, energy expenditure, and basal metabolic rate. It contributes to GDM by influencing adiposity and glucose usage. This network is highly regulated by the circadian clock, which may explain why pathological sleep disorders or those individuals undertaking shift work are correlated with GDM.35
Adipokines Involved in Gestational Diabetes Mellitus
Based on the evidence thus far, the adipokines adiponectin, leptin, TNF-α, and adipocyte fatty acid-binding protein (AFABP), seem to be the most probable candidates involved in the pathophysiology of GDM.
Leptin increases insulin sensitivity by influencing insulin secretion, glucose usage, glycogen synthesis, and FA metabolism. In skeletal muscle, it stimulates basal, but not insulin-stimulated, glucose uptake through the PI3K-dependent pathway and prevents the accumulation of lipids by stimulating FA oxidation through activating AMPK and, in turn, suppression of the activity of acetyl coenzyme-A carboxylase.36 In isolated hepatocytes, it causes significant reduction of
glucose production from different gluconeogenic precursors through PI3K-dependent activation of phosphodiesterase-3B (PDE3B).37 Leptin can suppress insulin secretion through several mechanisms, including the decrease of proinsulin messenger RNA levels in β-cells under high glucose concentrations; inhibition of glucose transport into β-cells; PI3K-dependent activation of PDE3B that leads to a reduction of cAMP levels and, in turn, suppression of protein kinase-A involved in the regulation of Ca2+ channels and exocytosis; and inhibition of the phospholipase-C/protein kinase-C pathway.38
Both obesity and pregnancy are characterised by leptin resistance associated with impaired leptin signalling in the hypothalamus.39 Although the reasons are not yet known, maternal leptin levels increase from the earliest stages of pregnancy, implying that the increases are not solely due to maternal weight gain. Leptin may contribute to the pathophysiology of GDM by effects related to appetite control, body weight and composition, energy expenditure influences, and direct influence on pancreas function.39 A cohort study has demonstrated that hyperleptinaemia at <16 weeks gestation was predictive of increased risk of GDM later in pregnancy and that each 10 ng/mL increase in leptin concentration was associated with a 20% increase in GDM risk, independent of maternal pre-pregnancy adiposity and other confounders; however, further prospective studies are required to determine predictive ability in GDM.40 The dysregulation of leptin metabolism and/or function in the placenta may be implicated in the pathogenesis of GDM. Nonetheless, there are conflicting reports regarding placental leptin expression in GDM.41,42
Adiponectin may improve insulin signal transduction via an increase of tyrosine phosphorylation of insulin receptors in skeletal muscle and by suppression of TNF-α, and can enhance insulin secretion by stimulating both the expression of the insulin gene and exocytosis of insulin granules.43 Additionally, it works by decreasing hepatic glucose production through AMPK.44 Adiponectin may activate PPAR-α, leading to increased FA oxidation, and reduced ectopic fat storage through inhibiting lipolysis in adipose tissue, thereby increasing insulin sensitivity.45
There is good evidence that adiponectin is lower in pregnancy and in GDM.46 Downregulation of adiponectin may predict GDM several months before diagnosis, independent of BMI and insulin sensitivity.47 Hypoadiponectinaemia persists post-partum with GDM and may contribute to progression to T2DM.46 Thus, it may play a key role in mediating IR and β-cell dysfunction in the pathogenesis of both T2DM and GDM. It is thought that TNF-α and other proinflammatory mediators secreted in GDM suppress the transcription of adiponectin, further aggravating chronic low-grade inflammation.39
Adiponectin expression in the placenta is differently regulated by various cytokines.48 This suggests its significance in adapting energy metabolism at the maternofetal interface. Lower adiponectin DNA methylation levels on the fetal and maternal side of placenta are associated with higher maternal glucose and adiponectin levels.49 Maternal adiponectin decreases fetal growth by impairing placental insulin signalling and reducing insulin-stimulated amino acid transport;50 therefore, decreased concentrations may contribute to fetal macrosomia in women with and without GDM.
TNF-α and IL-6
It remains controversial as to whether upregulation of TNF-α in GDM precedes or is a consequence of disease.39 TNF-α quantified at 11 weeks of gestation was not significantly associated with the risk of developing GDM in a study; however, only 14 cases and 14 controls were included.51 Placental gene expression of TNF-α, IL-1, and their receptors has been reported to be increased in GDM in some but not all studies.16,52 Additional analysis is required to clarify the role of TNF-α and other inflammatory cytokines as a predictor of GDM development independent of BMI.
Adipocyte Fatty Acid-Binding Protein
Cross-sectional studies have consistently found increased circulating AFABP concentrations in patients with GDM after adjustment for adiposity, IR, triacylglycerol, C-reactive protein (CRP), and associations with newborn size and adiposity.53,54 However, no prospective study has assessed whether baseline AFABP concentrations predict the risk of GDM. Furthermore, prospective studies and data for expression of AFABP in the placenta, visceral, or subcutaneous adipose tissue of patients with GDM have not been published.
Adipokines Probably not Involved in the Pathophysiology of Gestational Diabetes Mellitus
The evidence for visfatin, retinol-binding protein-4, vaspin, resistin, omentin-1, apelin, chemerin, progranulin, fibroblast growth factor 21, lipocalin 2, tissue inhibitor of matrix metalloproteinase-1, and zinc-alpha-2 glycoprotein is contradictory and/or lacking.55
Energy Storage
Maternal fat mass increases throughout the pregnancy, with accumulation of fat observed on the trunk.56 During pregnancy, appropriate expansion of adipose tissue is vital to support nutrient supply to the fetus. However, hypertrophic adipose tissue is associated with excess adiposity followed by elevated adipose tissue inflammation.7 The relationship between hypertrophic growth of adipose tissue and inflammation causes adipose tissue IR in the third trimester. This situation is exacerbated in obese pregnancies and those with GDM.57 Adipocyte hypertrophy and reduced adipocyte differentiation in GDM is accompanied by downregulated gene expression of insulin signalling regulators, FA transporters, and key adipogenic transcription factors, such as PPAR.57 Adipose tissue from obese patients with GDM expresses high levels of ectonucleotide pyrophosphate phosphodiesterase-1, correlating with GLUT4 expression, IRS-1 serine phosphorylation, and induction of adipocyte IR.58 The combination of IR and reduced adipocyte differentiation hinders the tissue’s ability to safely dispose of excess energy, contributing to gluco and lipotoxicity in other peripheral organs.
Adipose Tissue Inflammation
Expansion of adipose tissue and increased lipid deposition in adipocytes leads to the development of a more inflammatory adipocyte state, including the secretion of different inflammatory mediators which induce the recruitment of monocytes and/or their differentiation into proinflammatory M1-like macrophages. Once recruited, these macrophages secrete additional chemokines, initiating a feed-forward loop, potentiating the inflammatory response and impairing adipocyte function.59 Adipose tissue macrophages secrete TNF-α, IL-6, IL-1β, monocyte chemoattractant protein (MCP)-1, and macrophage-inhibitory factor. These factors induce IR either by diminishing insulin receptor tyrosine kinase activity, increasing serine phosphorylation of IRS-1, or through the STAT3–SOCS3 pathway, which degrades IRS-1.60 Their production is under transcriptional control of two inflammatory pathways: 1) c-Jun-N-terminal kinase (JNK)-activator protein 1 (AP1); and 2) inhibitor of NF-kB kinase (IKK).60 These pathways are initiated by almost all of the mediators implicated in the development of IR, including oxidative and endoplasmic reticulum stress, saturated FA, and inflammatory cytokines, highlighting their importance in the pathogenesis of disease. TNF-α, IL-1β, and pattern recognition receptors activate JNK and IKKβ/NF-kB through classical receptor-mediated mechanisms.60 NF-kB and AP-1 transactivate inflammatory genes, which can contribute to IR in a paracrine manner. JNK, a stress kinase that normally phosphorylates the c-Jun component of AP-1 transcription factor, has been shown to promote IR through the phosphorylation of serine residues of insulin receptor.60 Unlike JNK, IKKβ does not phosphorylate IRS-1 and causes IR through transcriptional activation of NF-κB. Impairment of insulin signalling due to chronic low-grade inflammation in adipose tissue further stimulates expression of genes.56
The relationship between pregnancy and inflammation is complex. The widely accepted belief that increased adiposity equates to increased maternal inflammation may not be as evident during pregnancy as in the non-pregnant state. GDM placentas have been shown to secrete fewer proinflammatory cytokines than healthy placentas.61 This suggests that, while chronic low-grade inflammation appears to be important in the pathogenesis of GDM, the relationship may not be straightforward. The elevation in plasma CRP, MCP-1, IL-6, and IL-1 receptor antagonist levels of overweight or obese women with increasing BMI in early-to-mid pregnancy was not evident towards the end of pregnancy.62 Therefore, the authors have speculated that increased adiposity during pregnancy is not associated with enhanced inflammation, as opposed to the widely held belief. Additionally, there is evidence, although still limited, implicating that the association between adipokines and GDM may be independent of adiposity measures and suggesting other pathways linking adipokines to the development of GDM, although BMI is not an accurate measure.40,41,63 Future studies using objective measures of adiposity, for example, dual-energy X-ray absorptiometry, may help to clarify the adipokines-GDM relations independent of adiposity.
Oxidative Stress
Pathologic pregnancies, including GDM, are associated with heightened oxidative stress due to overproduction of free radicals, leading to abnormal mitochondrial function, and impaired free-radical scavenging mechanisms.64 Reactive oxygen species inhibit insulin-stimulated glucose uptake by interfering with both IRS-1 and GLUT4. The activation of NADPH oxidase by lipid accumulation in the adipocytes is a potential mechanism shown to increase the production of TNF-α, IL-6, and MCP-1, and decrease the production of adiponectin.7 Interestingly, iron supplementation in already iron-replete women is associated with GDM, possibly as a result of increased oxidative stress.65 Homocysteine is also thought to contribute to GDM via oxidative stress. Exposure of β-cells to even small amounts of homocysteine results in impaired insulin secretion.66 A recent meta-analysis reported significantly higher homocysteine concentrations among women with GDM.67 Deficiencies and imbalances of B vitamins that are essential for homocysteine homeostasis are associated with GDM.66
Adipose Tissue-Derived Exosomal microRNA
Obesity and its related diseases influence the expression of exosomal microRNA, with significant upregulation of microRNA-23-b, microRNA-103-3p, and microRNA-4429.68 MicroRNA-23-b and microRNA-4429 activate the TGF-β and Wnt/β-catenin signalling pathways, causing obesity-related conditions.68 MicroRNA-103-3p, known to target the insulin receptor signalling pathway, was previously found to be downregulated in diabetes.69 Exosomes from hypoxic adipocytes show an enrichment of lipogenic proteins modulating lipogenic pathways in neighbouring adipocytes and pre-adipocytes thereby transferring characteristics of adipocyte dysfunction.70 In obese pregnancies, adipose tissue-derived exosomes may communicate with the placenta and change its function and therefore contribute to the development of GDM.
Gut Microbiome
There is emerging evidence that the gut microbiome may contribute to metabolic diseases. The gut microbiota composition of women with GDM, both during and after pregnancy, resembles the aberrant microbiota composition reported in non-pregnant individuals with T2DM and associated intermediary metabolic traits.71 A study of stool bacteria in women with previous GDM reported a lower proportion of the phylum Firmicutes and higher proportion of the family Prevotellaceae.72 Firmicutes appear to be relevant to pathogenesis of GDM independent of diet, although the mechanisms underlying this are unknown. Prevotellaceae may contribute to increased gut permeability regulated by tight junction proteins, such as zonulin. Increased ‘free’ serum zonulin is associated with GDM.73 Increased gut permeability is thought to facilitate the movement of inflammatory mediators from the gut into the circulation, promoting systemic IR.
CONCLUSION AND FUTURE DIRECTIONS
GDM is likely to be a result of a complex and variable interaction of genetic, environmental, maternal, and fetoplacental factors in an integrated manner. The molecular mechanisms by which these factors participate in the pathophysiology of GDM is currently a challenge. The conclusions drawn from most of the trials conducted are limited mainly due to the lack of statistical power and the controversial results obtained. Greater understanding of molecular mechanisms and their contribution to GDM is required to develop effective treatments and prevention strategies.