Abstract
One of the many extra-hepatic manifestations of hepatitis B virus (HBV) infections is vasculitis. While the classic HBV-associated vasculitis is polyarteritis nodosa, other vasculitides have been reported. The authors present an atypical case of acute HBV-associated vasculitis in a 57-year-old male with tobacco use disorder, characterised by extremity ischaemia, gangrene, splenic infarction, and positive proteinase-3 antibodies without sinopulmonary, gastrointestinal, or renal disease. The aggressiveness of the patient’s disease necessitated pulse-dose corticosteroids, cyclophosphamide, and two courses of plasmapheresis, and ultimately required multiple amputations of fingers and toes.
Key Points
1. Hepatitis B (HBV) is a chronic viral infection that has several extra-hepatic manifestations, one of which is vasculitis. HBV is classically associated with polyarteritis nodosa. Cryoglobulinemic vasculitis and antineutrophilic cytoplasmic antibody-associated vasculitis have also been reported in patients with HBV.
2. The authors present an atypical case of HBV-associated vasculitis, characterised by digital ischaemia, splenic infarction, complete occlusion of the bilateral ulnar arteries, and a strongly positive proteinase-3 antibody. Skin biopsy revealed perivascular inflammation with fibrinoid vessel wall necrosis but intact internal elastic lamina, as well as a fibrin-rich thrombus.
3. Based on features of polyarteritis nodosa, antineutrophilic cytoplasmic antibody-associated vasculitis, and thromboangiitis obliterans, the patient was treated broadly with pulse dose glucocorticoids, cyclophosphamide, plasmapheresis, therapeutic anticoagulation, and systemic vasodilation. The authors propose that HBV-associated vasculitis exists on a spectrum, requiring individualised treatment. Their case highlights the importance of reviewing biopsy results with a pathologist when there is clinical uncertainty.
INTRODUCTION
Although the exact pathophysiological mechanisms are still unclear, the association between viral infections and the development of autoimmune-mediated vasculitis has been well documented, especially with hepatitis B virus (HBV). HBV has been classically associated with polyarteritis nodosa (PAN), and to a lesser extent cryoglobulinemic vasculitis, with case reports of HBV-associated antineutrophilic cytoplasmic antibody (ANCA)-associated vasculitis (AAV).1-3 The causal relationship between HBV and vasculitis has been supported by the fact that antiviral treatments exert significant therapeutic benefits for concomitant vasculitis.4
Broadly, vasculitis is classified by the calibre of the main vessels involved: small, medium, and large. AAV, including microscopic polyangiitis, granulomatosis with polyangiitis (GPA), and eosinophilic GPA, can involve small or medium vessels. PAN mainly involves medium vessels. Thromboangiitis obliterans (TAO), or Buerger disease, also classically affects small-to-medium sized vessels, though it is almost exclusively limited to vessels in the extremities.
AAV is a spectrum of diseases that classically manifest with a pulmonary-renal syndrome, plus or minus sinus involvement. As the name suggests, most cases have positive ANCA antibodies, with proteinase-3 (PR3) antibodies corresponding with GPA, and myeloperoxidase antibodies corresponding with microscopic polyangiitis. Eosinophilic GPA has been variably associated with both antibodies, and is distinguished by the presence of eosinophilia and symptoms of atopy. PAN typically presents with purpura, tender erythematous nodules reminiscent of erythema nodosum, renal insufficiency from renal arteritis, mononeuritis multiplex, and mesenteric arteritis. Orchitis, coronary arterial obstruction, splenic infarctions, and ischaemic retinopathy also occur in variable rates.5 TAO is strongly associated with tobacco product use, and is characterised by Raynaud’s phenomenon; superficial thrombophlebitis; and digital ischaemia, with infrequent effects on arteries supplying the brain, heart, kidneys, or intestines.6
In this case presentation, the authors will discuss a 57-year-old male with tobacco use disorder, who presented with acute peripheral-predominant ischaemia in the setting of acute hepatitis B infection. The patient was subsequently found to have strongly positive PR3 antibodies, splenic infarctions, and complete occlusion of bilateral ulnar arteries, and then rapidly evolved into dry gangrene of multiple digits, requiring amputation.
CASE PRESENTATION
A 57-year-old White male, with a past medical history of Roux-en-Y gastric bypass surgery eight years prior, renal cell carcinoma status post-partial nephrectomy 8 years prior, provoked subsegmental pulmonary embolism with lower left extremity deep vein thrombosis 4 years prior, and a 12.5 pack-year history of tobacco use, initially presented with 2 weeks of worsening pain and swelling in their bilateral hands and feet, Raynaud’s phenomenon, blistering of the skin on bilateral lower extremities to an external hospital. Initial workup revealed moderately elevated liver enzymes, and the patient was subsequently found to have a newly diagnosed HBV, with a viral DNA load of over 145 million copies/mL. The patient denied recent blood transfusions, new sexual partners, or abdominal pain. The patient was started on entecavir, given symptomatic management, and discharged with close follow-up.
Five weeks later, the patient presented to the authors’ hospital with 1 week of coldness in their fingers and toes, with parasthesias and ischaemic changes extending to the proximal interphalangeal joints. Other than the pain in their hands and feet, the patient denied inflammatory arthralgias, myalgias, joint stiffness, sicca symptoms, gastrointestinal upset, changes in urination, epistaxis, focal weakness, shortness of breath, or systemic symptoms. The physical exam was notable for cool hands and feet, with duskiness and purpura of multiple fingers and toes (Figure 1), livedo reticularis of bilateral lower extremities, and weak radial and dorsal pedal pulses. The patient’s laboratory results were significant for markedly elevated inflammatory markers, normocytic anaemia, persistently elevated HBV viral DNA, strongly positive PR3 antibody (measured at 259 AU/mL on Day 2 and 276 AU/mL on Day 3, with a reference range of 0–19 AU/mL), and low positive anti-cardiolipin IgG and IgM antibodies. The patient’s renal function remained stable throughout their hospitalisation. Their cryoglobulins were negative on admission and 2 weeks later, with an unremarkable workup for systemic connective tissue diseases, including antinuclear antibody, Scl-70, complements, myeloperoxidase, rheumatoid factor, cyclic citrullinated peptide, Sjögren’s syndrome A, Sjögren’s syndrome B, β2 glycoprotein, and lupus anticoagulant.
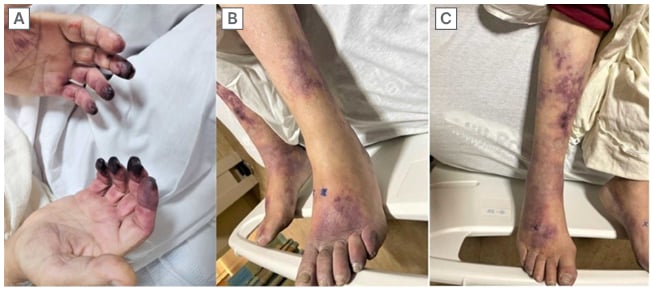
Figure 1: Dry gangrene on the tip of the digits (A) and toes (B), with purpura and livedo reticularis findings of the right lower extremity (C).
CT of the abdomen and pelvis with intravenous contrast revealed evidence of splenic infarctions that were not present on imaging 2 weeks prior (Figure 2A), which was confirmed with MRI imaging. CT angiography of the abdomen, pelvis, and lower extremities were negative for significant vascular abnormalities. Angiography of the bilateral upper and lower extremities showed complete obliteration of the patient’s ulnar arteries bilaterally beyond the wrist, with extraordinarily sluggish flow down both legs (Figure 2B and C).
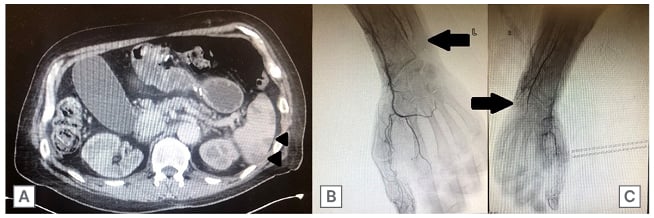
Figure 2: CT scans of the abdomen and pelvis, as well as the arms.
An axial cut of a CT scan of the abdomen and pelvis with intravenous contrast demonstrating numerous focal low-density areas in the spleen predominantly in the periphery (A; arrowheads), and an upper extremity angiogram, suggestive of distal occlusive disease (arrows) in the left arm (B) and right arm (C).
Skin biopsy of the left thigh revealed superficial perivascular neutrophilic infiltration, fibrinoid necrosis in vessel walls, and fibrin thrombus involving one medium size artery in the deep dermis with an intact internal lamina (Figure 3). Immunofluorescence was negative for IgG, IgM, C3, or fibrin immune deposits.
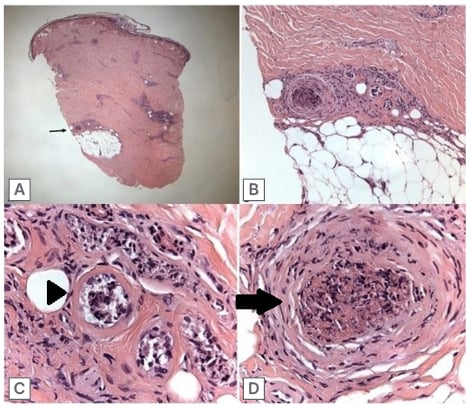
Figure 3: Skin biopsy.
A) 20x magnification (low power) of skin biopsy. The black arrow points to group of vessels of the most interest in the deep dermis/border of subcutaneous fat. B) 100x magnification of deep dermal vessels, showing a fibrin thrombus in a medium size vessel and a group of necrotic smaller vessels. C) 400x (high power) imaging of small necrotic vessels showing necrosis in the small vessels, with presence of fibrin (arrowhead). D) 400x magnification of the medium size vessel containing a thrombus (arrow), and showing some acute inflammation with nuclear debris in the vessel wall.
The patient was initiated on pulse dose glucocorticoids, therapeutic dose heparin, and vasodilation with nicardipine. Cyclophosphamide was started 5 days after admission at 600 mg/m2 every 2 weeks for a total of six doses. The patient also began plasmapheresis, with three sessions at the beginning of their hospitalisation. The patient eventually underwent amputation of second through fifth fingers bilaterally at the proximal interphalangeal joints, requiring revision amputation to the metacarpophalangeal joints, as well as trans-metatarsal amputation of the left foot. The patient’s course was complicated by gastric bypass anastomotic bleed, felt to be secondary to high dose steroids that, in combination with worsening skin changes, prompted their second course of plasma exchange for 5 days. The patient was informed explicitly about the potential use of their anonymised medical data and photographs for the sole purpose of medical research, teaching, and publication, and written consent from the patient was obtained.
DISCUSSION
HBV has several extra-hepatic manifestations, including vasculitis, among which the most common subtype is PAN. One large cohort study found 33.7% of cases of PAN were due to HBV, though more recent estimates put the frequency closer to 7% due to HBV vaccination efforts.1 Conversely, 1–5% of patients with HBV will develop PAN.7 Most cases of PAN develop within the first 6 months of HBV infection. Cryoglobulinemic vasculitis has also been reported in HBV, and occurs at a rate of 0.5–4.0% of patients who are infected with the disease.8-10 Multiple pathological mechanisms leading to endothelial damage of blood vessels have been suggested in HBV-associated vasculitis, including direct viral invasion, autoimmune antibodies formation, and immune complex deposition, and which may contribute to the complexity of its clinical manifestations.11,12 The authors’ patient had features of multiple vasculitides, including TAO, PAN, and AAV. Because of the overlap of characteristics, the authors’ therapeutic strategy was a combination of treatments directed at each distinct pathophysiology.
The vasculitis most classically associated with HBV infection is PAN. While the rate of PAN has decreased similarly with HBV, severe disease remains devastating, with up to 40% mortality at 5 years.13 The pathophysiology of HBV-associated PAN has been attributed to immune complex deposition.7 This is not always the case in patients without HBV, and a review article indicates that PAN exists on a spectrum, including adenosine deaminase 2 deficiency, as well as aberrant activation of the innate and adaptive immune systems.14 PAN can present with purpura, livedo reticularis, painful nodules similar to erythema nodosum, renal dysfunction from renal arteritis, abdominal pain from mesenteric arteritis, mononeuritis multiplex, and orchitis.15 Digital ischaemia, with or without necrosis, was reported in around 12% of patients with PAN.5 Splenic infarction is extremely rare in PAN, with only a handful of reported cases in the literature.16 In fact, the French Vasculitis Study Group (FVSG) did not report any cases with splenic involvement in their PAN database reviews.1,5 Since splenic infarction is typically silent, this finding could be under-reported, as discussed below.
The treatment of PAN depends on severity and HBV status. In mild cases of HBV-associated PAN, treatment is antiviral monotherapy. Entecavir is one of the most commonly prescribed antiviral agents for HBV. Common side effects include elevated liver function testing, headache, mild skin rashes, and haematuria; however, to date, it has not been implicated in the development of vasculitis. Those with severe HBV-associated PAN should also receive glucocorticoids and plasma exchange.17 Induction therapy for patients with mild systemic idiopathic PAN is glucocorticoids plus either methotrexate or azathioprine. Severe idiopathic disease should be managed with high-dose glucocorticoids plus cyclophosphamide.18
AAV is a necrotising vasculitis with a preponderance for small-to-medium vessels. The exact pathophysiology of AAV remains unclear, but ANCA have been shown to damage endothelium in vitro by activating both neutrophils and the alternative complement pathway.19-21 Patients can present with a wide variety of manifestations, including ocular inflammation, petechiae, purpura, respiratory distress from pulmonary infiltrates or diffuse alveolar haemorrhage, sinus disease (epistaxis, polyps, necrosis), renal dysfunction, and proteinuria or haematuria from glomerulonephritis. Digital ischaemia is very rare in AAV, with incidence of less than 1%, but is considered a severe disease manifestation in the American College of Rheumatology (ACR) management guidelines.22 Splenic infarction is likely underreported in AAV since the majority of patients are asymptomatic.23
The association between HBV and AAV is less clear. One study reported that 31% of patients with chronic HBV tested positive for ANCA, though the rate in the control group was 26%.24 They also demonstrated a higher rate of cytoplasmic ANCA and PR3 positivity as compared with perinuclear ANCA and myeloperoxidase in these patients with chronic HBV. Conversely, in a different cohort of patients with chronic HBV, only one out of 67 tested positive for ANCA.25 Association of HBV with AAV is even more limited, as the authors’ group was only able to find a single published case.3 In contrast, the risk of developing GPA or experiencing relapse has been correlated with chronic nasal carriage of Staphylococcus aureus.26,27
Management of AAV is separated into induction and maintenance phases. Induction therapy in mild cases without life or organ-threatening manifestations includes steroids with or without methotrexate, which is recommended over all other steroid-sparing agents.22 Those with severe disease, defined as having diffuse alveolar haemorrhage, glomerulonephritis, mononeuritis multiplex, cardiac involvement, mesenteric ischaemia, central nervous system disease, or limb/digit ischaemia, should be treated with high-dose steroids plus either rituximab or cyclophosphamide. The most recent ACR guidelines favour rituximab over cyclophosphamide based on demonstrated non-inferiority and better tolerability of rituximab.28 Plasma exchange should only be added for patients at high risk of progression to end-stage renal disease, concomitant anti-glomerular basement membrane disease, or as salvage therapy in refractory cases.22,29 After remission has been achieved, maintenance therapy should be initiated or continued. Those who had severe disease should receive rituximab for maintenance. Rituximab is favoured over methotrexate or azathioprine as rituximab was shown to have a lower relapse rate than azathioprine, and methotrexate and azathioprine have similar efficacy for remission maintenance.30,31 Patients who had mild disease are recommended to continue their same induction therapeutic agent.
Duration of maintenance therapy is unclear, but guidelines typically recommend a minimum of 18 months, with individualised decision making thereafter.
TAO does not seem to be associated with HBV, though its incidence has been correlated with periodontal infection.32 It is a segmental, inflammatory, non-atherosclerotic, occlusive vasculopathy. Tobacco product use is nearly ubiquitous in patients with TAO. Patients typically present with cold sensitivity of the fingers or toes, digital ischaemia, and superficial thrombophlebitis. TAO is an overwhelmingly peripheral disease, with very infrequent rates of cerebral, coronary, internal thoracic, renal, and mesenteric arteritis.6 Splenic infarction in TAO has only been described in three case reports.33-35 Histopathology reveals a highly cellular, inflammatory intraluminal thrombus with sparing of the vessel wall, and an intact internal elastic lamina.36
The cornerstone of treatment for TAO is smoking cessation, with emphasis on abstinence from both tobacco and cannabis. Since TAO is a vasculopathy, immunomodulatory therapy has not been shown to be efficacious in management, and the remainder of care is symptomatic, with some evidence supporting the use of vasodilators and intermittent pneumatic compression.37,38
The author’s case is unique, given that the patient had characteristics of all three vascular diseases. In one of the largest cohort trials of HBV-associated PAN, all patients (72 in total) who were tested for ANCA had negative results.1 In fact, ANCA positivity was an exclusion criterion in prior classification criteria for PAN.1 Since the aetiology of the patient’s condition was unclear, the authors implemented therapeutics that would work for all differentials considered. High dose steroids are central to the management of both PAN and AAV. Cyclophosphamide has also been shown to be effective in both diseases. Given the patient’s severe and refractory disease, plasma exchange was also an appropriate treatment modality. Smoking cessation and vasodilation were also implemented. Following the authors’ in-depth review of the case while preparing this manuscript, including discovery of an intact elastic lamina in the medium-sized cutaneous blood vessel weeks after discharge, the aetiology of this patient’s disease may well have been exclusively TAO. In the moment, it would have been difficult to justify withholding potentially limb-saving therapy given the patient’s HBV and PR3 positivity. Ultimately, this case serves as a reminder that treatment must be individualised, and based on a patient’s clinical status.
CONCLUSION
The authors present an uncertain and protracted case of HBV-associated medium vessel vasculitis with features of PAN, AAV, and TAO that required treatment with a combination of pulse-dose steroids, cyclophosphamide, and plasma exchange. The patient ultimately required amputation of multiple fingers and toes after demarcation and stabilisation of the vasculitic process. Given the lack of clarity surrounding the patient’s case, the authors propose that HBV-associated vasculitis is a heterogeneous disease that warrants an individualised therapeutic strategy, based on disease manifestations and severity.






