Abstract
Russell first described the diencephalic syndrome in 1951. It is a rare syndrome and usually presents in children as a cause of failure to thrive despite normal, or even increased appetite, with preservation of linear growth. The treatment options vary from endoscopic biopsy followed by chemotherapy to definitive surgical resection of the tumour. The authors here describe a case of an 8-year-old 10 kg emaciated child who presented with headache, vomiting, rage attacks, decreased weight, and diminution in vision. The child had bilateral optic atrophy; however, hormonal profiles were within normal limits. MRI of the brain gave an impression of craniopharyngioma.
Thus, the child was planned for subtotal resection of the tumour. There were various anaesthetic concerns in this case. The low weight predisposed them to hypothermia, electrolyte abnormalities, titrating the dose of drugs accordingly, positional challenges due to fixed flexion deformity of limbs, and the need for extensive post-operative care. Post-operatively, the child developed diabetes insipidus 2 hours after surgery, which was medically managed, but the repeat episode occurred again the next day. On second post-operative day, the child succumbed to dyselectrolemia and thrombocytopenia, despite resuscitative measures. The literature available on the anaesthetic management for definitive surgical resection is scarce, especially the post-operative complications encountered in this case. Thus, this case report could help in elaborately understanding the spectrum of the disease, especially regarding this histopathological type with its associated complications and, therefore, the steps that could be taken to mitigate them.
Key Points
1. Children with the diencephalic syndrome are prone to poor prognosis due to their severely emaciated state and associated complications; hence, vigilant screening in failure to thrive children could help early diagnosis before landing in such an emaciated state.2. The histological subtype of craniopharyngioma further adds to the anticipated complication in forms of diabetes insipidus, which could be challenging to manage in these syndromic patients.
3. Tailoring the peri-operative anaesthetic management to the specific considerations in these cases and providing appropriate post-operative care is crucial for better surgical outcomes for such patients.
INTRODUCTION
Diencephalic syndrome (DS), first described by Russell in 1951, is a rare cause of failure to thrive in infants and young children with preservation of linear growth.1 Due to misdiagnosis of these children, these conditions are diagnosed quite late and, by then, the children already would be in a state of severe emaciation. This delay makes them candidates for poor prognosis, even when they get definitive treatment. Most of the literature available for this rare syndrome focuses on the chemotherapy aspect of the treatment. The literature on anaesthetic considerations and post-operative complications after surgical the resection of these brain tumours is scarce. This mode of therapy is usually not opted for considering their anatomical location, which could make complete resection difficult. But since the tumour in the indexed case was so large, the debulking of the tumour was chosen to increase the efficacy of planned post-operative chemotherapy. The authors of this report elaborate on the anaesthetic management and post-operative complications of diabetes insipidus and thrombocytopenia in an emaciated 8-year-old male child, weighing 10 kg, with suprasellar mass posted for debulking and subtotal excision of the tumour.
CASE REPORT
Clinical Findings: History and Physical Examination
An 8-year-old male child, weighing 10 kg (Z score: -12.29), presented with a complaint of headache for 2 years, which was gradually progressive and mainly involved the frontal region. There was also a history of vomiting for 6 months, drowsiness for 2 months, and increased head size and diminution of vision for the past month. The parents also gave an account of rage attacks and decreasing weight despite increased appetite, but no history of fever, seizures, or trauma.
About 1 month previously, the child became drowsier and, at that time, they were diagnosed with hydrocephalus on radiological imaging. A ventriculoperitoneal shunt was performed with the uneventful post-operative course. The patient was referred to the authors’ tertiary care centre for further management. On examination, the child was conscious and co-operative with the Glasgow coma scale (GCS) of eye opening (E) 4; verbal response (V) 5; and motor response (M) 6. Their visual acuity was severely reduced, with the right eye’s perception of light but the perception of light was absent in the left eye. Bilateral optic atrophy was diagnosed on fundoscopy. There was fixed flexion deformity at bilateral elbows, hips, and knees, with the power of three out of five in all the limbs. The child’s low weight and fixed flexion deformity of the limbs had crucial significance as the dosage of the drugs should be modified as per the lean bodyweight of the child, and positioning of the child should be done with utmost care with proper cushioning of the pressure points.
Diagnostic Assessment
Biochemical work-up
Routine investigations were all within normal limits. The initial haemoglobin was 11.5 g/dL, leading to a maximum allowable blood loss of 110 mL. The baseline platelet count was 3.87×103 cells/µmm; growth hormone 2.45 ng/mL (normal: up to 3 ng/mL); adrenocorticotrophic hormone 13.4 pg/mL (normal: up to 46 pg/mL); and thyroid function tests (triiodothyronine: 6.5 ng/dL; thyroxine: 6.65 µg/dL; thyroid-stimulating hormone: 1.53 µIU/mL) and cortisol (8.4 µg/dL) were within normal limits. The endocrinologist’s opinion was taken, and they suggested that since all the hormones were within normal limits, no additional supplementation in the form of steroids and thyroxine was required.
Radiological Evaluation
MRI of the brain suggested that a well-defined lobulated solid cystic mass, measuring 4.5×3.5×5.5 cm, involving the sellar and suprasellar regions was causing expansion of the sellar (Figures 1 and 2). Superiorly, the lesion was uplifting and compressing the optic chiasma and also abutting and displacing the A1 segment of bilateral anterior cerebral artery. Inferiorly, the lesion was compressing the pituitary gland. Laterally, the lesion abutted bilateral cavernous sinuses, with the normal bilateral internal carotid artery. Posteriorly, it was compressing the pons, midbrain, and basilar artery, splaying the mid-brain and cerebral peduncles. It was also compressing the third ventricle and aqueduct of Sylvius, causing gross dilatation of bilateral lateral ventricles with an Evan’s Index (EI) of 0.62 and the radiological impression of craniopharyngioma. Considering the tumour’s proximity to neurovascular structures, invasive modalities such as invasive arterial blood pressure monitoring and central venous cannulation were planned.
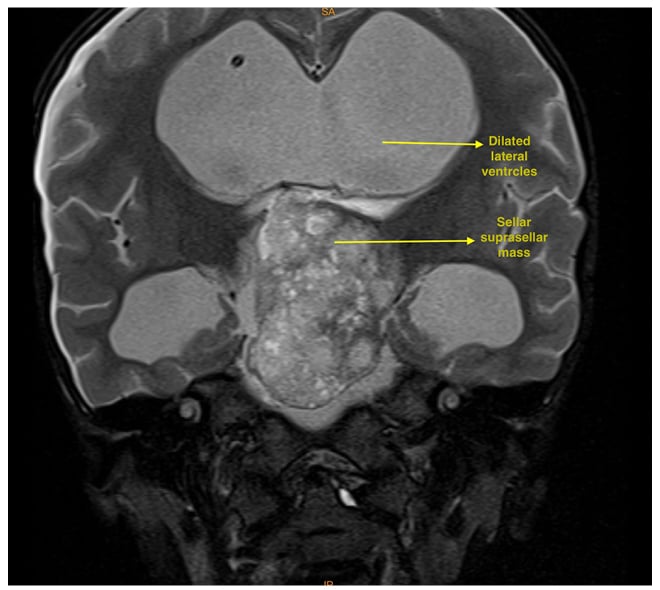
Figure 1: T2 coronal MRI showing a dilated lateral ventricles and large hyperintense sellar and suprasellar mass.
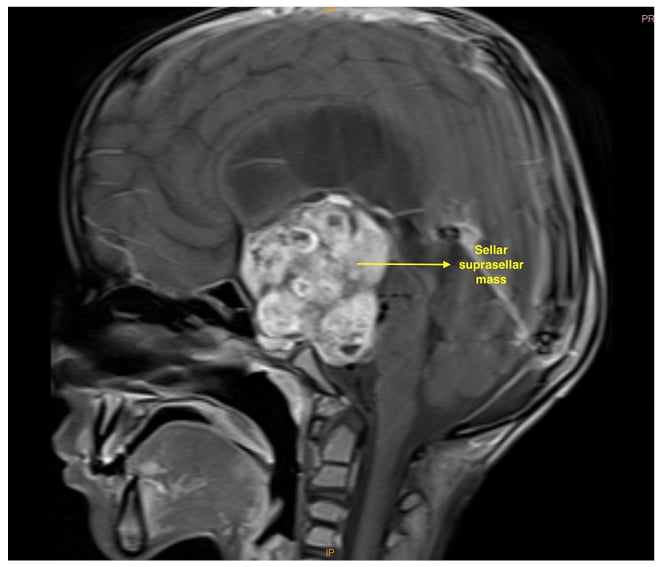
Figure 2: T1 sagittal MRI showing a large hypertense sellar and suprasellar mass.
Therapeutic Intervention
Given the large size of the tumour, chemotherapy would not have been very effective. Hence, the patient was planned for left pterional craniotomy and debulking of the tumour under general anaesthesia, which was to be followed later by chemotherapy.
Intraoperative Anaesthetic Management
The pre-operative assessment, including airway examination, was typical, with a Modified Mallampati score of 1. After confirming nil per oral status on the day of surgery, the patient was taken to the operating theatre. Their baseline non-invasive blood pressure was 110/75 mmHg, pulse rate 102 /min, and oxygen saturation 99% on room air. Proper positioning with cotton padding of all pressure points was completed to prevent peripheral nerve injury. The forced-air warming blanket was used to avoid hypothermia, and the operating room’s temperature was maintained at 25 °C. The intraoperative temperature was monitored using a skin temperature sensor and adjusted for core temperature. Pre-medication with injection glycopyrrolate 0.05 mg was done. Intravenous induction was achieved with an injection if fentanyl 20 µg and propofol 20 mg. Muscle relaxation was achieved with an injection of vecuronium 1 mg and intubated with a cuffed endotracheal tube sized 5.0 mm without difficulty. Maintenance of anaesthesia was performed with oxygen and air (40:60), sevoflurane 1 minimum alveolar concentration, and intermittent boluses of vecuronium and fentanyl. Ventilation was done to maintain end-tidal carbon dioxide 33–5 mmHg. The right dorsalis pedis artery was cannulated for the beat-to-beat blood pressure monitoring. Right subclavian central venous access was done with a 5.5 Fr triple lumen central venous cannula. As preoperative cortisol levels were within normal limits, intraoperative steroids were not supplemented.
Surgical Management
Intraoperatively, the surgeon observed that the pituitary gland and stalk could not be separately identified, and optic apparatus was splayed and thinned out. The tumour was infiltrating the hypothalamus and abutting the brain stem. Hence, the surgeon decided to leave the posterior attachment of the tumour on the brainstem and the remaining part of the tumour was removed; thus, haemostasis was achieved. The total duration of anaesthesia was 8 hours. Intraoperatively, blood loss of 300 mL was replaced with 150 mL packed red blood cell. Intraoperative arterial blood gas and blood sugars were within normal limits. Normothermia was maintained throughout the intraoperative period. The patient was shifted to the neurological intensive care unit for elective ventilation and delayed extubation.
Follow-Up and Outcomes
Urine output increased (120 mL /hour for 4 hours) 2 hours post-operatively. The anticipated post-operative complication of craniopharyngioma, diabetes insipidus, was suspected and serum electrolytes were sent. Hypernatremia was detected, with postoperative sodium levels tabulated in Table 1. There were no other electrolyte derangements. However, serial measurements of urine osmolality and serum osmolality would have been a better predictor for diagnosis and prognosis. Still, they could not be done as they were unavailable at the authors’ institute at that time.
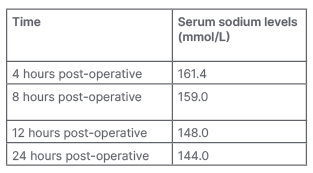
Table 1: Serial serum sodium levels.
Free water (50 mL every 3 hours) was administered through a nasogastric tube and 0.45% of normal saline intravenously, 30 mL per hour. An injection of desmopressin 0.05 mg was administered after consultation with an endocrinologist. Over the next 4 hours, urine output started decreasing appropriately. On the first post-operative day, they were extubated as the child gained full consciousness, responded to commands, and had intact airway reflexes.
A repeat episode of increased urine output and hypernatraemia occurred after 24 hours. The injection of desmopressin 0.05 mg was repeated but, gradually, the patient’s GCS started deteriorating to E3V3M4. The patient was put on mechanical support again. The specimen’s histopathological biopsy confirmed the craniopharyngioma diagnosis (World Health Organization [WHO] Grade 1).
On the second post-operative day, the patient developed new-onset nystagmus, their GCS further decreased to E2V non-testable M3, and they developed haemodynamic instability with hypotension. They were started on an injection noradrenaline infusion at 0.1 µg/kg/min and titrated according to blood pressure. A complete blood count revealed an unanticipated complication of thrombocytopenia with a 1,4000 /µmm platelet count, and four random donor platelet transfusion units were done. A non-contrast CT brain scan showed no sign of haemorrhage. Unfortunately, despite all the resuscitation measures, the patient succumbed to dyselectrolemia due to diabetes insipidus and thrombocytopenia the next day.
Timeline of Events
In December 2019, the child started having a frontal headache, which progressed in intensity over 2 years. From June 2021, the child developed vomiting, rage attacks, and decreased weight despite increased appetite. In October 2021, their parents noticed drowsiness in the child. From November 2021, the child complained of diminution in vision. At that time, on evaluation, he was diagnosed with hydrocephalus, and a ventriculoperitoneal shunt was performed. In December 2021, child was brought to our tertiary care centre for further management. They were clinically examined, and it was found that they had a low weight for their age (Z score: -12.29), and there was bilateral optic atrophy. The patient was investigated thoroughly with blood investigations, including hormonal profiles, which were within normal limits. Radiological evaluation with MRI brain gave an impression of craniopharyngioma, and they were planned for sub-total resection of the tumour. Post-operatively, the child developed diabetes insipidus 2 hours after surgery, which was medically managed; however, a repeat episode occurred again the next day. It was further complicated by the onset of thrombocytopenia. The child succumbed to dyselectrolemia due to diabetes insipidus and thrombocytopenia on the second post-operative day.
DISCUSSION
An 8-year-old child with loss of weight, average linear growth, and the absence of gastrointestinal symptoms and other neurological signs had a diagnosis made favouring DS.2 DS is associated with various types of tumours of the hypothalamic and optic chiasmatic region, the most common being low-grade glioma followed by astrocytoma.3 However, in this case, the histopathology was craniopharyngioma (WHO Grade 1).
The usual age of presentation is 18 months in an emaciated child in the presence of average linear growth despite normal or even increased appetite. These children attain typical milestones as per age and appear to be more than happy for their state, which could be because of personality changes owing to the tumour itself. The Boston Children’s Hospital, Massachusetts, USA, and the Dana–Farber Cancer Institute, Boston, Massachusetts, USA, showed the incidence of hyperkinesia (9%), nystagmus (27%), and emesis (36%).3
The pathophysiology of this rare disease is still unclear, although increased growth hormone and ghrelin, as well as decreased insulin and leptin levels, have been suggested. However, these hormonal changes were claimed to be related to the patient’s nutritional status rather than causality.4,5 This patient had a typical hormonal profile, although the tumour was close to the hypothalamus and pituitary gland. The possible explanation for this finding could be that the tumour had not compressed the pituitary gland enough to alter the hormonal profile.
The survival rate without treatment after the onset of symptoms is 12 months.6
The treatment can vary from chemotherapy after the endoscopic biopsy to definitive surgical excision of the lesion. The concept of the HIT-LGG-1996 study recommends early chemotherapy instead of radiotherapy in younger children with low-grade gliomas.7 Conventional frontline therapy for patients with DS consists of chemotherapy, typically with agents like carboplatin and vinca alkaloids.8 Kim et al.9 conducted a retrospective study of 8 patients with DS in 2015. Three patients received concurrent chemoradiotherapy, two received chemotherapy, and three did not receive treatment. On follow-up evaluation, two patients were still receiving chemotherapy, and three patients showed improvement and remained in stable remission.
Radiotherapy is not chosen, given the endocrinological and neurological consequences in paediatric patients. The most recent case reported was by Roncall et al.10 in 2019, of case of DS in an 8-year-old child posted for endoscopic biopsy and ventriculoperitoneal shunt under general anaesthesia.10 The child was discharged home after the uneventful post-operative course, with the initiation of chemotherapy.
The treatment protocol for DS has not been standardised to date, and no clear guidelines are available on which treatment can be based. The protocols are usually chosen based on the specific type of tumour causing DS. Chemotherapy appears to play a significant role in the management of DS in young children.11
As this was suspected as craniopharyngioma, considering the WHO guidelines for new onset craniopharyngiomas. Sub-total resection of the tumour was planned first as it was a large tumour. Hence, debulking the tumour using surgical resection would help increase the success of postoperative chemotherapy for residual tumour.
Anaesthetic considerations specific to this case were: positioning with padding of all the pressure points due to loss of subcutaneous fat and fixed flexor deformity of joints; risk of hypothermia due to loss of subcutaneous fat and postoperatively due to hypothalamic injury; drug dosing and fluid management as per the weight of the child; massive fluid shift, electrolyte imbalance, and haemodynamic instability due to diabetes insipidus; and caloric requirements of the child to be met post-operatively.
The post-operative thrombocytopenia that the authors encountered in the case study presented here could be dilutional in aetiology due to a massive fluid shift due to diabetes insipidus, as the patient was given free water via nasogastric tube as well as 0.45% normal saline intravenously. This was supported by the finding of thrombocytopenia on blood haemogram, despite normal platelet count pre-operatively. Although correction of hypernatremia was given as per the free water deficit calculation, the patient showed an exaggerated response, which manifested as an overcorrection of their serum sodium levels (Table 1). The complication of diabetes insipidus is quite common after craniopharyngioma surgery. Still, it was more detrimental here as the child was severely emaciated, and thus had a lower percentage of total body water and could not cope with the diabetes insipidus. Therefore, a timely diagnosis and case-tailored management in such cases are essential before the stage of emaciation sets in. The post-operative neurocritical care in these patients is targeted on an illiberal fluid replacement to prevent fluid overload, management of diabetes insipidus, preventing hypothermia, meeting caloric requirements, preventing bed sores due to pressure necrosis, and guided ventilation to prevent respiratory complications.
To the best of the authors’ knowledge, no case has been reported where diabetes insipidus has been observed in patients with the diencephalic syndrome. Thus, this case report can help further understand the spectrum of disease and its complications.
LIMITATIONS
There were a few limitations in this case study. The child presented with emaciation due to delayed diagnosis, which increased the probability of complications. Also, the massive size of the suprasellar tumour also increased the chances of complications.
Parents’ Perspective
The parents did not know that their child had a brain tumour for a long time. They saw their child losing weight daily in front of their eyes, and they saw their child start to develop new symptoms as the days passed; however, they could not find the reason for it and kept wondering if there was hope of finding treatment. After coming to the authors’ hospital, the patients found out that their child had a brain tumour. The delay in getting therapy could have been one of the reasons for the untimely death of their child. Had they received a timely diagnosis and management, maybe their child could have been saved. The doctors and staff did their best to manage their child, but things did not turn up in their favour and parents lost their child.