
Abstract
Barrett’s oesophagus (BO) is one of the most important complications of gastro-oesophageal reflux disease as it may progress to oesophageal adenocarcinoma. There is currently a rising incidence of BO. The pathogenesis of BO is not well-understood although genetic and environmental factors play significant roles. BO can be dysplastic or non-dysplastic. In the case of dysplastic BO, two pathologists’ opinions are required. As patients with BO can be entirely asymptomatic, it is difficult to screen this population group. Currently, screening is recommended only for white males with certain risk factors according to American College of Gastroenterology (ACG) guidelines in the USA. The extent of BO can be reliably measured using the Prague classification. Patients with non-dysplastic BO should have surveillance endoscopy every 3–5 years, whereas dysplastic BO should be ablated endoscopically. Any nodule in the BO should be removed by endoscopic mucosal resection. Patients with BO should be on life-long acid-suppressant therapy. Non-invasive techniques such as the Cytosponge are being investigated as an alternative to endoscopy for BO screening.
INTRODUCTION
Barrett’s oesophagus (BO) is one of the most common gastrointestinal conditions gastroenterologists manage in their clinical practice.1 It is named after the Australian-born British thoracic surgeon Norman Barrett who first recognised the condition in 1950. This is an acquired condition that occurs as a result of chronic injury to the oesophagus due to gastro-oesophageal reflux disease (GORD). During the healing process, any part of the normal lining of stratified squamous epithelium in the distal oesophagus is replaced by a metaplastic simple columnar epithelium with mucin secreting goblet cells, also called specialised intestinal metaplasia (SIM).2 The columnar mucosa is easily recognised endoscopically by its salmon-pink colour. At the present time in the USA, BO is defined as an extension of salmon-coloured mucosa into the distal oesophagus ≥1 cm above the gastro-oesophageal junction (GOJ) with histological confirmation of SIM. SIM with goblet cells is the sine qua non for the diagnosis of BO. The British Society of Gastroenterology (BSG) defines BO slightly differently, as an extension of salmon-coloured mucosa above the GOJ with histological confirmation of columnar mucosa. SIM is not a requirement in this definition. The main importance of BO is that it is a precursor to oesophageal adenocarcinoma (OAC) although the risk is low.3
EPIDEMIOLOGY
The exact prevalence of BO in the general population of the USA is unknown as it is present in both symptomatic and asymptomatic patients. The majority of BO patients are asymptomatic and therefore the condition is under-diagnosed. BO is found in 10–15% of symptomatic GORD patients when their upper endoscopy is done.4 In one study, the prevalence of BO was found to be 5.6% in patients who were undergoing colonoscopy and never had any heartburn.5 A simulation model confirmed by the US Surveillance Epidemiology and End Results (SEER) cancer registry data showed the estimated prevalence of BO in the general population to be 5.6%.6 A population-based study in Sweden found the prevalence of BO to be 1.6%.7 If we apply this estimate to the USA, then 3.3 million people >50 years of age have BO. BO is mostly seen in the sixth or seventh decades of life, the average being 55 years, but it can develop at an earlier age. In a Dutch study there was a 20-year age shift in the development of BO.8 Men developed the disease 20 years earlier than women. As a result, the prevalence of BO in the young population had a male to female ratio that approached 4:1. Overall the disease is twice as common in males than in females.9 The annual incidence of BO is highest among non-Hispanic white populations and much lower in Hispanic, Asian, and Black populations.10 With the epidemic of GORD, the incidence of BO has increased over the last few decades.11
RISK FACTORS
There are certain risk factors for the development of BO in patients with GORD:
- Age: middle-aged to elderly
- Sex: male
- Race: Caucasian
- Chronicity of GORD symptoms for >5 years. The longer the duration of GORD symptoms, the higher the chance of developing BO.12
- Central obesity (in males, waist circumference >102 cm or waist-hip ratio >0.9; in females, waist circumference >88 cm or waist-hip ratio >0.8) which significantly increases the risk of developing BO.13 One study showed that central obesity was strongly associated with BO, especially long-segment BO, in the White population but not in Hispanic or Black populations.14
- Cigarette smoking which contributes to the development of BO (a modest risk factor) in GORD patients.15
- Hiatus hernia which is associated with the development of BO, particularly long-segment BO.16
- Family history: germline mutations in MSRI, ASCCI, and CTHRCI genes have been associated with the development of BO. Familial forms of BO account for 7–11% of all cases.17
- Obstructive sleep apnoea, an independent risk factor associated with increased incidence of BO.18
- Metabolic syndrome which is associated with an increased risk of BO irrespective of symptoms of GORD.19
- Low birth weight. A population-based study in Sweden showed that infants born small for gestational age had a 3-fold increased risk of developing BO as adults compared with infants of normal birth weight.20
PROTECTIVE FACTORS
- Gastric colonisation of CagA-positive strains of Helicobacter pylori may protect against the development of BO.21
- Intake of vegetables and fruits may reduce the risk of developing BO in both males and females.22
Intake of non-steroidal anti-inflammatory drugs does not reduce the risk of developing BO.23
PATHOGENESIS
The exact pathogenesis of SIM occurrence in the distal oesophagus is not known. GORD is considered to be the initiating factor.24 It is proposed to be a two-step process.25 In the first step, there is a transformation of squamous epithelium into simple columnar epithelium. With continued acid exposure there is injury to the distal oesophageal mucosa and dilated intracellular spaces are seen histologically.26 This increases mucosal permeability and allows particles ≤20 kDa to reach the oesophageal stem cells in the basal layer. In the reparative process, the oesophageal stem cells differentiate into columnar cells instead of squamous cells. BMP4 is overexpressed in inflamed oesophageal mucosa and BO.27 In ex vivo models, BMP4 induces transformation of squamous mucosa to columnar mucosa. In the second step, intestinal metaplasia occurs by more than one cellular pathway. Expression of intestinal genes allows goblet cells to populate in the columnar epithelium leading to intestinal metaplasia. The CDX2 gene plays an important role in this transformation process.28
There is a moderate-to-strong CDX2 protein expression in the Barrett’s mucosa. It has been shown to be associated with the development of intestinal metaplasia from columnar epithelium. The CDX promoter is activated on exposure to acid and bile,29 thus duodenogastro-oesophageal reflux of bile may also play a role. Bile in the acidic pH becomes non-ionised and soluble. It can enter the oesophageal epithelial cells and can cause toxic injury to mitochondria and cellular mutations.30 A speculative sequence linking different molecular pathways has been proposed. BMP4 expression in the oesophageal mesenchyme is first upregulated by pro-inflammatory agents such as acid and bile. Stem cells in the basal layer of oesophageal epithelium are then activated. There is activation of gene transcription leading to columnar metaplasia. The CDX2 gene is then activated with the formation of intestinal metaplasia.31 The columnar mucosa extends up into the distal oesophagus either as tongues or circumferentially.
There are several theories about the cell of origin of BO.32 These include migration of stem cells from gastric cardia, migration of embryonic population at the GOJ, migration of bone marrow stem cells, upward migration of submucosal gland cells, and transdifferentiation of squamous epithelial cells into columnar epithelial cells. An acid pocket at the GOJ may also contribute to the pathogenesis of BO. Dietary nitrate is reduced to nitrite by buccal bacteria. Nitric oxide is then produced at the GOJ acid pocket. Nitric oxide can cause metaplasia and neoplasia.33
PATHOLOGY
In non-dysplastic BO (Figure 1), the squamous mucosa is replaced by columnar mucosa with mucin-filled blue goblet cells. Nuclei are regular and aligned basally. Glands are well spaced and surrounded by plenty of lamina propria. Dysplastic, i.e. intra-epithelial neoplastic, BO is characterised by lack of cytoplasmic maturation, nuclear changes, and crowding of glands. In low-grade dysplasia (LGD) there is cytological atypia up to the mucosal surface (Figure 2), nuclear atypia with hyperchromatism and pleomorphism, increased mitosis, and crowding of glands with decreased intervening lamina propria, but nuclear polarity is preserved. In high-grade dysplasia (HGD), there are marked cytologic changes (Figure 3): enlarged, hyperchromatic, and pleomorphic nuclei with at least focal loss of polarity, increased and atypical mitosis, and crowding of glands with almost complete loss of lamina propria.34
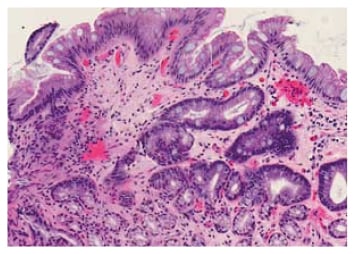
Figure 1: Non-dysplastic Barrett’s oesophagus (haematoxylin and eosin stain).
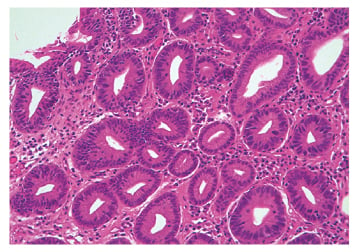
Figure 2: Low-grade dysplasia (haematoxylin and eosin stain).
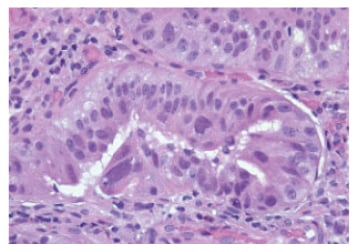
Figure 3: High-grade dysplasia (haematoxylin and eosin stain).
CLINICAL FEATURES
Patients generally have a long-standing history of heartburn or acid regurgitation but they can be completely asymptomatic. Sometimes they may present with dysphagia because of development of OAC or Barrett’s ulcer.
DIAGNOSIS
BO is most often suspected during upper endoscopy, showing salmon-coloured and coarsely textured mucosa extending from the GOJ into the distal oesophagus lined by smooth light pink or white squamous mucosa. The salmon-coloured mucosa represents columnar mucosa and the top of the gastric folds marks the GOJ. Normally, the diaphragmatic hiatus/pinch, GOJ, and squamocolumnar junction (Z-line) remain at the same level. The location of these landmarks should be recorded during endoscopy. In the case of BO (Figure 4) there is displacement of the Z-line ≥1 cm above the GOJ. Endoscopic grading for BO was developed and validated in the Prague classification: the circumferential length (Prague C) and maximal length (including tongues) of salmon-coloured mucosa (Prague M) are measured in cm.35 This classification is very practical as Barrett’s segment can be tracked over time using this classification. Although patchy islands of Barrett’s mucosa are not mentioned in the Prague classification, they should be included in the endoscopy report. The extent of Barrett’s mucosa is dependent on the severity of the gastro-oesophageal acid reflux.36
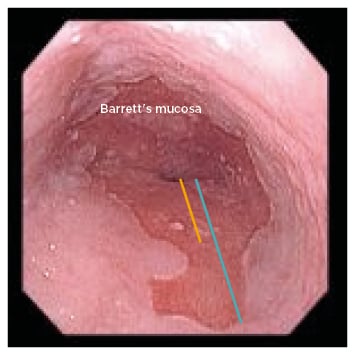
Figure 4: Endoscopic picture showing salmon-coloured mucosa with a tongue in the distal oesophagus suggestive of Barrett’s oesophagus. Prague C (yellow) and M (blue) are also shown at an estimated 2 and 4 cm, respectively.
During endoscopy, at least eight random biopsies (four quadrant biopsies every 2 cm) should be taken from suspected Barrett’s mucosa to increase the yield of SIM. In cases of suspected short segment BO (<3 cm) where eight biopsies cannot be taken, at least four biopsies should be taken per cm of circumferential suspected Barrett’s mucosa, and one biopsy per cm in tongues of suspected Barrett’s mucosa. If there are any visible lesions such as mucosal irregularities, nodules, or ulcers seen, targeted biopsies should be taken prior to random biopsies. To minimise the obscured field from bleeding, biopsies should be taken distally first and then proximally. Endoscopic biopsy should not be taken in cases of irregular Z-line or if the Z-line is <1 cm above the GOJ1 as intestinal metaplasia of the GOJ and gastric cardia is quite common in clinical practice;37 it is mainly associated with H. pylori infection and is not a risk factor for OAC.38 If the biopsy is negative in suspected Barrett’s mucosa, endoscopy should be repeated in 1–2 years. In one study, 29% of patients had SIM in 2 years following initial negative endoscopic biopsy.39
SCREENING
The main purpose of screening is to detect patients with BO who can benefit from the surveillance programme. BO is the transition phase between GORD and OAC. As mentioned earlier, a large number of patients with BO can be completely asymptomatic. One study showed that 39% of patients with OAC did not give any history of long-standing heartburn or regurgitation.40 On the other hand, the risk of OAC in non-dysplastic BO is very low.41 Currently, there are no controlled trials showing that screening for BO in the general population is cost-effective. Nonetheless, screening and surveillance for BO in a high-risk group can be cost-effective.42 The American College of Gastroenterology (ACG) recommends screening for BO in males with chronic (>5 years) and frequent (weekly or more) symptoms of heartburn or acid regurgitation, as well as ≥2 of the following risk factors for BO or OAC:
- Aged >50 years
- Non-Hispanic white population
- Central obesity
- Present or past history of smoking
- Confirmed family history of BO or OAC in a first-degree relative
There should be a low threshold to screen for BO in the absence of multiple risk factors if the patient has a positive family history of BO or OAC as per BSG guidelines. Screening for BO in females is not recommended by ACG because of the significantly lower risk of OAC in females. If multiple risk factors as mentioned are present in males, screening should be considered in individual cases. Although screening for BO in females is not routinely recommended by the ACG, the BSG still advocate for it.
Instead of sedated standard endoscopy, unsedated ultrathin transnasal endoscopy (TNE) with topical anaesthetic can be used for screening for BO. As the thin scope goes through the nose without touching the base of the tongue there is no gag reflex and sufficient tissue biopsy can be taken from Barrett’s mucosa. One study showed TNE was not only accurate, it caused less anxiety in patients.43 Although the ACG accepts TNE as an alternative to standard endoscopy for BO screening, the BSG does not recommend it currently because of insufficient evidence. If the first screening for BO is negative, subsequent endoscopy screening for BO is not recommended. If oesophagitis (Los Angeles classification B, C, or D) is seen during screening, the patient should receive 8–12 weeks of proton pump inhibitor (PPI) therapy for the healing of oesophagitis, followed by a repeat endoscopy. The overall life expectancy of the patient should be considered prior to screening for BO. The patient should be informed about the subsequent programme of endoscopic surveillance and therapy in cases of BO with dysplasia.
As endoscopy is an expensive and invasive procedure, different non-invasive techniques are being investigated for BO screening. One of them is the Cytosponge, a small (30 mm in diameter) sponge made of meshes and covered by a soluble gelatine capsule, and attached to a string. When patients swallow the sponge, the gelatine coating dissolves in the stomach. After 5 minutes, the sponge is taken out by pulling the string.44 The oesophageal cells are collected from the sponge and immunohistochemistry is examined for TFF3, a marker of SIM. The sensitivity and specificity of the test has been found to be 73.0% and 93.8%, respectively,45 however because of its lower sensitivity, screening with the Cytosponge needs to be validated in future. TFF3 along with different predictive, prognostic, and progression biomarkers are currently being studied for risk stratification,46 however they are not yet ready to be used in clinical practice.
SURVEILLANCE
The main objective of surveillance of patients with BO is to detect dysplasia or OAC at an early stage so that effective treatment can be given. One study showed that endoscopic surveillance detected BO-associated OAC at a lower stage and there was an improved survival.47 The surveillance programme starts with endoscopic biopsy of the Barrett’s mucosa: four quadrant biopsies at 2 cm intervals in patients with non-dysplastic BO, and four quadrant biopsies at 1 cm intervals in patients with known dysplastic BO. High definition white light endoscopy should be used. The ACG does not support routine use of advanced imaging except narrow-band imaging (NBI), which is a form of electronic chromoendoscopy. NBI may help in detecting dysplasia if targeted biopsies are taken from areas with irregular pattern.48 The BSG does not recommend either chromoendoscopy or virtual chromoendoscopy for surveillance of BO.49 Risk stratification of BO using biomarker panels is not recommended either by the ACG or BSG. The efficacy and cost-effectiveness of endoscopic surveillance versus at-need endoscopy is now being studied in the multicentre randomised controlled BOSS trial.50
CHROMOENDOSCOPY AND BARRETT’S OESOPHAGUS
Chromoendoscopy involves topical application of dye or stains on the oesophageal mucosa during endoscopy and has been clinically available for many years. However, due to lack of consistent use in clinical practice it has never become very popular. There are different forms of chromoendoscopy with different staining patterns. Lugol’s iodine gives a brownish to almost black stain of the normal squamous mucosa of the oesophagus as it binds with glycogen in squamous epithelium. BO does not stain with Lugol’s iodine. Methylene blue binds with the intestinal epithelium but not squamous mucosa of the oesophagus, thus enhancing detection of BO. Indigo carmine stains both squamous and columnar mucosa. As it lies in surface pits and groves, it gives an excellent topographic map of the mucosa delineating subtle changes so that targeted biopsy can be taken. Toluidine blue has affinity for mucopolysaccharides and thus can stain goblet cells seen in BO. Acetic acid (1.5%) enhances the visibility of mucosal surface borders and texture, thus helping to map out the Barrett’s mucosa for targeted biopsy. Chromoendoscopy has been used in the evaluation and surveillance of BO.51
Electronic Chromoendoscopy or Dye-Less Chromoendoscopy
Image enhancement or colour alteration of the image can be conducted by manipulating the wavelength of the light source or digital post-processing. This technology is available in Olympus scopes as NBI, in Pentax scopes as i-SCAN, and in Fujinon scopes as Fujifilm Intelligent Color Enhancement (FICE). Enhanced view of the texture of the mucosal surface and blood vessels can be seen by this technology. NBI is considered as optical chromoendoscopy, whereas i-SCAN and FICE are virtual chromoendoscopy techniques.
MANAGEMENT
Depending on the histology of surveillance biopsies, further actions are taken:
- Non-dysplastic BO: endoscopic surveillance should be done every 3–5 years as per ACG. BSG takes into consideration the length of BO for surveillance interval. Patients with BO <3 cm should have surveillance endoscopy every 3–5 years, whereas BO ≥3 cm should be surveyed every 2–3 years.
- Indefinite for dysplasia: patient should receive PPI therapy for 3–6 months. Repeat surveillance endoscopy with biopsies should then be done. If the histology is again indefinite for dysplasia, endoscopic surveillance should be done after 12 months.
- Dysplastic BO: any grade of dysplasia should be reviewed and confirmed by two pathologists, of whom at least one should be a gastrointestinal pathologist.
- LGD: endoscopic ablative therapy is the first choice in the absence of life-limiting comorbidity. Alternatively, endoscopic surveillance every 12 months is acceptable.
- HGD or intramucosal carcinoma (IMC): endoscopic ablative therapy is the treatment of choice in the absence of life-limiting comorbidity. Any nodule or mucosal abnormality should be removed by endoscopic mucosal resection (EMR).
Radiofrequency ablation (RFA) using BARRX™ RFA (Medtronic, California, USA) is considered the most effective and preferred modality of endoscopic ablation therapy and is now the standard of care in the USA. RFA is safe and highly effective in eradicating both intestinal metaplasia and dysplasia in BE.52 Post-RFA therapy, nausea, chest pain, and dysphagia can occur temporarily, but the oesophageal stricture rate is approximately 1–6%.52,53
NODULAR BARRETT’S OESOPHAGUS
EMR should be done for both diagnostic and therapeutic purposes if there is any discrete nodule in the Barrett’s mucosa. If the histology of the nodule (Figure 5) shows:
- HGD or IMC: the rest of the Barrett’s mucosa should be treated with endoscopic ablative therapy.
- Tumour invades lamina propria or muscularis mucosa (T1a) OAC: endoscopic ablative therapy of the remaining BO is the next step.
- Tumour invades submucosa (T1b) OAC: endoscopic ultrasound is usually carried out to evaluate the depth of tumour infiltration, and to biopsy local lymph nodes as there is a high rate of lymph node involvement in T1b OAC.54 Oesophagectomy with consideration of neoadjuvant therapy is the preferred treatment. Endoscopic ablative therapy is an alternative if the carcinoma is superficial (sm1), well-differentiated without any lymphovascular invasion, or if the patient is a poor surgical candidate.
- T1b, sm2–3 OAC: oesophagectomy with consideration of neoadjuvant therapy is the preferred treatment.
- Irrespective of T1a or T1b OAC, if there is any poor differentiation of carcinoma, lymphovascular invasion, or incomplete EMR, surgery with neoadjuvant therapy should be considered.
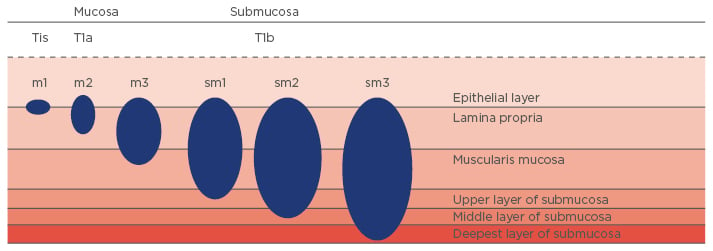
Figure 5: Early oesophageal adenocarcinoma staging.
Tis: tumour infiltration into mucosal epithelial layer (carcinoma in situ); T1a: tumour invades lamina propria or muscularis mucosa; T1b: tumour invades submucosa; m1: tumour infiltration into mucosal epithelial layer; m2: tumour infiltration into mucosal lamina propria; m3: tumour infiltration into muscularis mucosa; sm1: tumour infiltration into the upper third of submucosal layer; sm2: tumour infiltration into the middle third of submucosal layer; sm3: tumour infiltration into the deepest third of submucosal layer.
The European Society of Gastrointestinal Endoscopy (ESGE) strongly recommends endoscopic submucosal dissection if the Barrett’s nodule or lesion is >15 mm, poorly lifting, or at risk of submucosal invasion.55
ENDOSCOPIC ABLATION
Circumferential or focal RFA is performed56 and then repeated after 3–4 months if there is residual intestinal metaplasia. On average, two to three treatment sessions are required over a period of 4–12 months to achieve complete elimination of intestinal metaplasia. Endoscopic surveillance is required after complete elimination of intestinal metaplasia. The oesophagus and GOJ should be examined using white light and NBI in antegrade and retrograde views. The protocol of surveillance should be every 3 months in the first year, every 6 months in the second year, and then annually thereafter in cases of HGD or IMC. In cases of LGD, endoscopic surveillance should be done every 6 months in the first year and then annually. During surveillance, if there are any recurrent metaplasia and/or dysplasia, they should be managed as per the treatment options mentioned earlier.
As BO is a complication of GORD, adequate acid suppression for an indefinite period of time is required as part of the treatment. Patients should be on PPI therapy regularly for controlling acid reflux symptoms as well as for the healing of the reflux oesophagitis. Currently the effectiveness of aspirin and esomeprazole for the prevention of OAC in patients with BO is being studied in an ongoing Phase III clinical trial, AspECT.57
SUMMARY
With the epidemic of GORD and more awareness among gastroenterologists, BO is becoming increasingly recognised in clinical practice. GORD patients with certain risk factors are more prone to developing BO. Although there are speculations as to how BO is developed, the precise pathogenesis is unknown. Patients with BO have 30 to 40-times increased risk of developing OAC than those without BO.58 There has been >7-fold increased incidence of OAC over the last few decades. The approximate annual risk of developing OAC is 0.12–0.50% for non-dysplastic BO and 6% for dysplastic BO.59 One study showed that patients with non-dysplastic BO developed LGD at 4.3% per year and HGD at 0.9% per year.60 Another study showed that 26.5% of patients with LGD developed HGD or OAC over a 3-year period.61 As OAC is a deadly disease, screening has been suggested in individuals with multiple risk factors. Patients screened positive for BO should be under a surveillance programme to detect dysplasia or OAC at an early stage. The ACG recommends endoscopic surveillance every 3–5 years for non-dysplastic BO because of a low risk of developing dysplasia or neoplasia in this group, and endoscopic ablation therapy for dysplastic BO. EMR should be done for any discrete nodule seen in the Barrett’s mucosa. A nodule with T1a OAC can be managed endoscopically by EMR and ablation of the Barrett’s mucosa. Oesophagectomy is recommended for nodules with T1b OAC. All post-ablation therapy patients should be on a surveillance programme. All patients with BO should be taking life-long acid-suppressant therapy, preferably a PPI once a day. Patients should be counselled to help them understand the benefits and risks of screening and surveillance programmes.






