Abstract
Nonalcoholic fatty liver disease (NAFLD) has become an emerging disease throughout the world. Metabolic comorbidities such as obesity (especially central obesity), diabetes, and dyslipidaemia have been established as risk factors not only for NAFLD development, but also for the disease progression. Dietary or genetic obesity has been hypothesised to induce alteration of gut microbiota, thereby causing the promotion of deoxycholic acid production in the intestinal tract. Elevated levels of deoxycholic acid can provoke senescence-associated secretory phenotype in hepatic stellate cells through enterohepatic circulation, which in turn leads to the secretion of various inflammatory and tumour-promoting factors in the liver and may further result in obesity-induced hepatocellular carcinoma. Short-chain fatty acids are mainly produced through the fermentation of indigestible carbohydrates by gut microbiota. Gut microbiota have been considered to play a role in NAFLD and its disease progression. The main end products resulting from the indigestible carbohydrate catabolism of intestinal microbes are short-chain fatty acids, constituting acetate, propionate, and butyrate. High concentrations of propionate can promote development of NAFLD, whereas acetate and butyrate can prevent the development of the disease.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is commonly associated with metabolic comorbidities such as obesity, diabetes, and dyslipidaemia. For diagnosis of NAFLD, the patient must have evidence of hepatic steatosis, either from imaging or histology, and lack of significant alcohol consumption (<20 g/day) or any other agents that contribute to liver fat accumulation. NAFLD is now considered as the third most common cause of hepatocellular carcinoma (HCC) in the USA.1
Gut dysbiosis is an imbalance condition of gut bacteria which has been hypothesised to have an important role in NAFLD. Short-chain fatty acids (SCFA) are mainly produced by the fermentation of indigestible carbohydrates (CHO) by gut microbiota.2The exact role of SCFA in the pathogenesis of NAFLD is still unclear. In this review, the nature of gut microbiota, the role of SCFA, and their implications on NAFLD are discussed.
NATURAL HISTORY, PREVALENCE, AND RISK FACTORS OF NONALCOHOLIC FATTY LIVER DISEASE
NAFLD is a wide spectrum of diseases developed from simple steatosis, potentially progressing to steatohepatitis and then to liver cirrhosis or HCC development. Based on its histopathology, NAFLD can be differentiated into two phenotypes: the usually stable condition, nonalcoholic fatty liver (NAFL), without hepatocellular injury evidence; or nonalcoholic steatohepatitis (NASH) with evidence of hepatocyte injury. Most patients with NAFLD will remain in a stable condition. If the liver histopathology only shows isolated steatosis or simple steatosis, patients will usually have a benign prognosis; however, some of the NAFLD population will progress to NASH. Prevalence of NASH ranges between 6% and 13%, and >50% attain condition stability or go into regression. Approximately 9–20% of the NASH population will progress to cryptogenic cirrhosis and 40–60% of them will experience cirrhosis-related complication, including HCC, over 5–7 years.3,4
Studies have shown that most of the metabolic parameters such as obesity, hyperlipidaemia, hypertension, and diabetes are strongly related to NAFLD.5-10Similar small studies in Asia conducted by Lesmana et al.11and Amarapurkar et al.12showed several metabolic components that were significantly associated with disease progression in NASH patients. The metabolic components strongly associated with NASH included obesity, hyperlipidaemia, and Type 2 diabetes mellitus (DM). Based on a World Health Organization (WHO) dataset, there was increased prevalence of overweight and obese individuals in 2016, and this is predicted to rise further in the future.4Based on follow-up research conducted in Indonesia, the prevalence of obesity (BMI >27.0 kg/m2) in adults has significantly increased in the last 12 years, from 10.5% in 2007 to 21.8% in 2018.13
In line with this, there is an increased prevalence of NAFLD in the world, particularly in the USA where it has ranged between 15% and 25% for 5 years (Republic of Indonesia Ministry of Health, data on file).Indonesia, the largest country in south-east Asia, has also shown the clinical importance of NAFLD prevalence. The first Indonesian study conducted by Hasan et al.14 in 2002 revealed that from 808 subjects in a community based in Depok, 30.6% experienced NAFLD. An additional study conducted by Lesmana et al.15in 2015, showed from a patient database that 51% of adult NAFLD cases are initially found in transabdominal ultrasound check-ups in hospitals.
These two studies also analysed the risk factors of NAFLD development. The first study conducted by Hasan et al.14identified the statistically significant risk factors for NAFLD as age, triglyceride level, DM, and obesity, whereas sex, fat intake, and physical activity were not associated with NAFLD. Lesmana et al.15 conversely found that sex, age, blood pressure, BMI, fasting blood glucose, triglycerides, high-density lipoprotein, and serum alanine transaminase were statistically significant risk factors for NAFLD development. From these two studies, it can be concluded that NAFLD is a hepatic manifestation of the metabolic syndrome family, and obesity is considered as the most important factor for NAFLD based on the insulin resistance mechanism. This conclusion has been supported by many studies from Western countries.16,17
GUT MICROBIOME
The human microbiome exists in many parts of the body and outnumbers human cells by a ratio of 10:1. Gut microbiota play a key role in nutrition, fighting against pathogenic bacteria, and influencing developmental factors of the body, from body weight to brain development. Development of the gut microbiome begins at birth and continues to adulthood. The first phase of development of the gut microbiome is determined by whether a baby is born naturally or by caesarean section. In the next phase, the baby’s gut microbiome can be influenced by nutritional intake: especially by breast or formula milk. Breast feeding has an important role in increasing immunity, whereas formula milk has a role in increasing Bacteroides, Clostridium, and Clostridium difficilebacteria. Additionally, beneficial bacteria in the gut may decrease if the individual has a history of frequent antibiotic treatment.18
After the breastfeeding period, the baby will start to eat solid food. In this period, Firmicuteswill develop as a sign of the adult gut microbiome ecosystem forming. The last stage of gut microbiome development is influenced by daily food intake. The traditional diets that contain high fibre and CHO will help develop more Prevotellain the human gut, whereas a Western diet which typically contains high fat will develop more gut Bacteroides. The imbalance of diversity and function of intestinal microbiota might contribute to disease development: either intestinally, as with inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), and coeliac disease; or extraintestinally, with obesity, DM, and metabolic syndrome.19-21
Features of Gut Microbiota in Obesity
Some alterations of gut microbiota in obesity will be associated with many factors, such as satiety control in the brain; hormone release from the gut (shown as peptide YY and GLP-1); synthesis, storage, or metabolism of lipids in the adipose tissue, liver, and muscle; and intestinal permeability related to systemic inflammation and insulin resistance. Microbial molecules also increase intestinal permeability, leading to systemic inflammation and insulin resistance (Figure 1).22
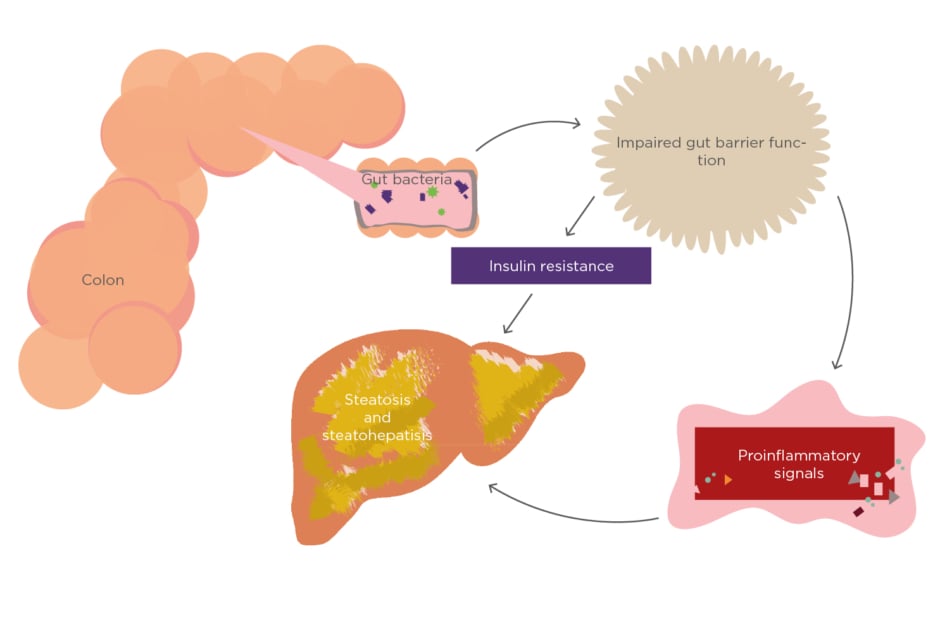
Figure 1: The role of gut microbiota in nonalcoholic fatty liver disease.
The Role of Gut Microbiota in Nonalcoholic Fatty Liver Disease and Disease Progression
Gut microbiota are mostly belonging to phyla such as Firmicutes, Bacteroidetes,Actinobacteria, Proteobacteria, Verrucomicrobia, and Fusobacteria. The main phyla dominating the gut are Firmicutesand Bacteroidetes. The disruption of the gut microbiota composition leads to the imbalance condition, termed gut dysbiosis.23
In high free fatty acid accumulation conditions such as obesity, obesity itself can cause gut dysbiosis and lead to insulin resistance, and it has been shown to have a strong association with NAFLD development. Gut dysbiosis has also been hypothesised to have a role in liver disease progression.23 Studies have shown that small intestinal bacterial overgrowth (SIBO), which is a condition of gut dysbiosis, is strongly related to NAFLD development. There is, however, ongoing debate surrounding the data on this phenomenon.24-26
Research conducted by Yoshimoto et al.27 analysed the gut microbiota in two groups of leptin-deficient mice (lean and obese mice) and treated them with a chemical carcinogen. After the intervention, the mice fed with a high-fat diet had more Gram-positive bacteria and significantly higher serum deoxycholic acid levels, a gut bacterial metabolite known to cause DNA damage. The obese mice eventually developed HCC. In the study, the model of obesity-induced HCC was proposed from the DCA-SASP (senescence-associated secretory phenotype) axis. Alterations of gut microbiota in mice were induced by dietary or genetic obesity, thus increasing the levels of DCA. DCA in enterohepatic circulation provokes SASP in hepatic stellate cells. This mechanism induces secretion of various inflammatory and tumour-promoting factors in the liver, which facilitate HCCdevelopment in mice after chemical carcinogen exposure. This hypothesis has also been supported by results from mice lacking a SASP inducer or depletion of senescent hepatic stellate cells which efficiently prevents development of HCC.21 HCC development was also influenced by the activation of various cell signalling pathways, e.g., EGFR-pathway,28 ERK1/2-pathway,28AKT-pathway,29 andβ-catenin pathway,30 by DCA.
This hypothesis is also supported by the recent study by Fitriakusumah and Lesmana,31which shows that in NAFLD patients, the prevalence of metabolic factors was dominated by obesity (76.6%), followed by dyslipidemia (75.2%), central obesity (73.2%), metabolic syndrome (73.0%), and DM (70.0%), where SIBO was found to be higher in patients with NAFLD (65.5%) than those without NAFLD. Although there is no significant association between SIBO and NAFLD development, further analysis in the study showed that in the significant fibrosis group, there was a positive correlation to the presence of Bacteroides(p=0.037).31 Based on the Asia-Pacific Working Party on NAFLD guidelines 2017,32 it is stated that overnutrition, insulin resistance, and obesity are still the most important factors in NAFLD development. The most important risk factors for gut microbiota are largely still based on experimental data.
GUT MICROBIAL SHORT-CHAIN FATTY ACIDS AND THE METABOLIC PATHWAY
The main end products resulting from CHO catabolism of intestinal microbes are known as the faecal metabolites, specifically SCFA which include acetate, propionate, and butyrate. The gut microbiota are also the producers of these SCFA. The genus of gut microbiomes known as the primary source of dietary fermentation are Roseburia, Ruminococcus, Salmonella, Blautia, Eubacterium,Anaerostipes, Coprococcus, Faecalibacterium, Marvinbryantia, and Megasphaera. Most of these genera belong to the phyla Firmicutesor Bacteroidetes.23,33
Acetate and propionate are produced by Bacteroidetesphylum, whereas butyrate is mostly produced by the Firmicutesphylum. Acetate is the most abundant SCFA in the colon and has two main metabolic routes: from pyruvate via Acetyl-CoA and via the Wood–Ljungdahl pathway (reductive Acetyl-CoA pathway). Three different pathways are used by colonic bacteria for propionate formation: the succinate pathway, acrylate pathway, and propanediol pathway. In contrast, butyrate has two different pathways: the butyrate kinase and butyryl-CoA pathway. Each of these pathways have their own specific bacteria as producers of SCFA (Figure 2).34
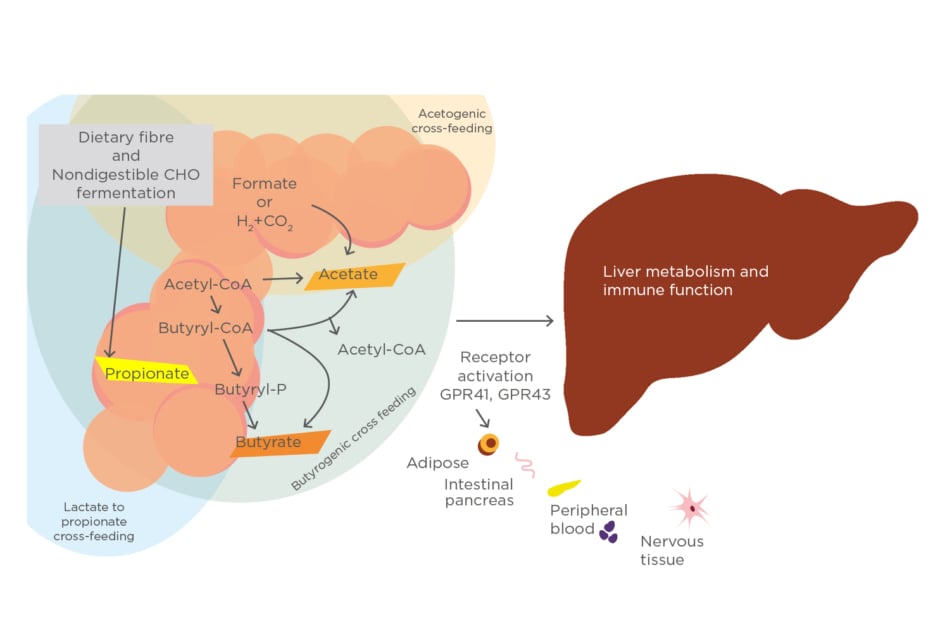
Figure 2: Microbial metabolic pathways to short-chain fatty acids in the human gut.
CHO: carbohydrate.
THE NATURAL MECHANISM OF SHORT-CHAIN FATTY ACIDS AND FOOD RELATED ISSUES
The receptors for SCFA are of the G-protein-coupled class (GPCR). These receptors are found in various organs, meaning that SCFA can affect multiple parts of the body. Receptors for SCFA include GPR43, GPR41, and GPR109. GPR41 and GPR43 are activated by acetate, propionate, and butyrate. Meanwhile, GPR109 is activated by butyrate and niacin. The SCFA transporter is also found in the colonic epithelium. Transporters important for the handling of SCFA by the colon include monocarboxylate transporter 1 (MCT1) and sodium-coupled monocarboxylate transporter 1 (SMCT1) in the apical membrane, and MCT1 and MCT4 in the basolateral membrane. MCT1, a H+-coupled low-affinity transporter, works under normal dietary conditions to allow SCFA entry into the colonic epithelium when the luminal fibre concentration is high. In contrast, SMCT1, a Na+-coupled high-affinity transporter, plays a critical role under low luminal fibre conditions as a consequence of low fibre content in the diet. SCFA transporters, therefore, provide beneficial effects for colonic bacteria, allowing them to produce their fermentation product in the colon.35
Dietary CHO is usually absorbed in the intestine and delivered to the liver through the portal vein. Fast food and food containing high sugar (simple CHO) are not recommended for routine consumption because they are directly absorbed and induce de novolipogenesis (DNL), causing liver fat accumulation which will disturb the balance of SCFA concentration. Fermented and high-fibre foods (complex CHO) are strongly recommended for consumption because gut microbiota use nonfermented CHO (fibre) as raw materials in their metabolism to produce SCFA. This is because dietary fibre cannot be digested in the intestines, and therefore the fermentation process is performed in the colon. Fermented food can also be used as a source of both probiotics and prebiotics.36 Dietary fat, largely consumed as triglycerides, is also very important as it is usually coming after lipolysis. Saturated fat is more dangerous than unsaturated fat because it is more easily accumulated in the liver and can cause hepatocyte injury. It also has the potential to cause hepatic steatosis and an insulin resistance condition.37
EFFECT OF SHORT-CHAIN FATTY ACIDS ON NONALCOHOLIC FATTY LIVER DISEASE
SCFA has been considered as one of the key parameters in the pathogenesis of NAFLD. It is known that SCFA has some impact on adipose tissue, energy metabolism, the immune system, and also has a role as the gut microbiota-derived signalling mediators. The role of SCFA in NAFLD starts after its absorption process and travels through the portal vein into the liver. It has been shown from experimental studies that T-cell regulation and neutrophil chemotaxis can be influenced by SCFA.36,38
Based on natural SCFA mechanism, liver fat is regulated mainly by receptor activation (GPR41, GPR43, GPR109A, and OLFR78). The GPR43 receptor is known to be related to the three SCFA: acetate, propionate, and butyrate. In studies this receptor has been shown to be increased in mice receiving a high-fat diet. High abundance of SCFA will lead to hepatic steatosis, meanwhile decreased SCFA reduces hepatic lipogenesis.36,39
Acetate is known to be involved in insulin sensitivity and plays a role in modulating gut hormones. However, butyrate and propionate are more important in preventing carcinogenesis, maintaining the intestinal barrier, preventing inflammation, and are also influential in satiety regulation. Acetate plays an important role in lipogenesis and cholesterol synthesis in the liver as well as in the peripheral tissue. Acetate can contribute to the accumulation of triglycerides, and propionate has been known to be involved in hepatic lipid metabolism. Both acetate and propionate are also responsible for low inflammation levels through the immune cellular system. In a probiotic study using the strain Clostridium butyricum, butyrate prevented liver disease progression by reducing lipid accumulation in the liver and improved insulin resistance.36
SCFA can also promote NAFLD development through secretion of peptide YY and GLP-1; however, butyrate is the only SCFA which is hypothesised to prevent NAFLD through decreasing both the immune response and insulin resistance.36 An experimental study investigated the role of GLP-1 and showed that butyrate promoted this receptor, and that there was an apparent reduction of this receptor in NAFLD patients.40Another recent study in Europe used faecal collection analyses and showed that NAFLD patients with significant liver fibrosis had low levels of faecal acetate and propionate concentrations when compared to healthy controls. There was also a positive correlation between faecal SCFA (propionate/acetate) and peripheral proinflammatory T cells, supporting the evidence for the role of SCFA in liver disease progression in NAFLD/NASH.41
CONCLUSION
Gut microbiota plays an important role in NAFLD development and disease progression. SCFA faecal concentrations may be used as NAFLD biomarkers for disease prediction in the future. High concentration of propionate can promote the development of NAFLD, whereas acetate and butyrate can prevent the development of NAFLD.