
Meeting Summary
Biosimilars follow a rigorous regulatory approval pathway designed to collect and review the totality of evidence from non-clinical analytical comparability exercises as well as clinical Phase I and III studies between the biosimilar and the reference biological. Once the European Medicines Agency (EMA) has given a positive opinion on the generated totality of evidence, the agency may extrapolate the biosimilar’s clinical data from the indication in which the biosimilar was studied to other indications for which the reference biological was approved. A prerequisite for this step is a convincing demonstration of biosimilarity within a studied clinical Phase III population that is suitably sensitive to detect potential clinically relevant differences in efficacy, safety, or immunogenicity. This regulatory pathway was used for all currently available biosimilars including SB2 (Flixabi®), a recently approved biosimilar that is licensed for use across all indications approved for its reference biologic infliximab (Remicade®), including inflammatory bowel disease (IBD). Further to robust non-clinical evaluations of SB2 in 46 physicochemical and 23 biological assays, a Phase I study demonstrated pharmacokinetic equivalence between SB2 and reference infliximab. Furthermore, a Phase III study performed in patients with moderate-to-severe rheumatoid arthritis (RA) — a scientifically appropriate, sensitive patient population — showed that SB2 was equivalent to infliximab in terms of its primary endpoint, American College of Rheumatology 20% improvement (ACR20) response rate at Week 30, and comparable with regard to safety and immunogenicity up to Week 54. Additional analyses of treatment-emergent adverse events (TEAEs) by anti-drug antibody (ADA) status up to Week 54 demonstrated a comparable incidence of TEAEs in both treatment arms. The ACR response rates, safety, and incidence of ADAs remained comparable also in the transition extension period up to Week 78 between patients who continued to receive either SB2 or reference infliximab, and patients who transitioned from reference infliximab to SB2. Biosimilars have an important place in the treatment of IBD. Increased use of biosimilars in patients with Crohn’s disease (CD) or ulcerative colitis is likely to reduce costs, expand access of eligible patients to biologic therapy, and improve overall health outcomes.
Introduction
Anti-tumour necrosis factor (TNF) monoclonal antibodies (mAbs) may induce sustained clinical remission and mucosal healing, reduce the need for steroids, and may limit the progression of IBD.1-4 As some currently available anti-TNF mAbs reach patent expiry, similar versions of these reference products — known as biosimilars — are entering the market. Indeed, two infliximab biosimilars (CT-P13 and SB2) are now licensed for the treatment of IBD in Europe. Biosimilars have the potential to offer substantial cost savings compared with their reference products, potentially expanding patient access to effective IBD treatments and improving overall health outcomes.5
Clinicians, patients, and their pharmacists need to understand that biosimilars follow a different regulatory approval pathway to that used for novel (the biosimilar ‘reference’ or ‘originator’) products. Pioneered by the EMA, the approval pathway for biosimilars focusses primarily on establishing analytical comparability between the biosimilar and the reference product.6 Furthermore, if biosimilarity is demonstrated clinically in a sensitive patient population, the EMA may consider appropriate scientific justification to extrapolate these data to other indications for which the reference biologic is approved, without asking for additional clinical trials in these corresponding indications.6-8 This concept of extrapolation is especially relevant for gastroenterologists, because anti-TNF biosimilars have largely been evaluated in non-IBD populations. There are reasons for this, as will be discussed.
SB2 (Flixabi®) is an infliximab biosimilar that has been developed by Samsung Bioepis Co., Ltd (South Korea), a joint venture between Samsung BioLogics and Biogen. SB2 is licensed for use in the European Union (EU) across all indications approved for reference infliximab: RA, adult and paediatric CD, adult and paediatric ulcerative colitis, ankylosing spondylitis (AS), psoriatic arthritis, and psoriasis. In this Biogen-sponsored, interactive symposium held at the United European Gastroenterology (UEG) Week 2016, internationally recognised specialists used the example of SB2 to discuss the development process required by the EMA for biosimilar approval and how biosimilars are being introduced into IBD treatment algorithms.
Regulatory and Developmental Pathways of Biosimilars: An Innovative Concept
“[For me], it was very important to understand the regulations of the EMA in terms of biosimilars when I was considering my decision whether or not to use these drugs in my clinical practice.” Prof Julián Panés
The EMA defines a biosimilar as “a biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product.”6 As a result of the inherent complexity of biologically-derived proteins, the development of biosimilars requires a different approach to that used with chemically-derived small generic molecules. The innovative regulatory pathway followed by the EMA involves a comprehensive comparability exercise, with the aim of demonstrating similarity to the reference product in terms of physicochemical characteristics, mode of action and non-mode of action-related biological activity, clinical safety, immunogenicity, and efficacy.6 The primary amino acid sequence, pharmaceutical form and strength, indications, and route of administration for the biosimilar need to be the same as for its reference product.
The regulatory approach adopted by the EMA is based on the scientific principles applied when evaluating the impact of post-approval manufacturing changes on the characteristics of originator biologicals.6 Indeed, it is important to realise that these originator biologicals are almost always, themselves, a different version of the originally licensed product. Such changes vary widely in their likelihood to affect the structural and functional attributes of the reference biological, ranging from a change in filter supplier (low-risk) to a new cell line, major formulation change, or implementation of new manufacturing sites (high-risk).9 For example, reference infliximab (Remicade®) has undergone 50 post-approval changes, 13 considered to be low-risk, 14 moderate-risk, and 3 high-risk.9,10 Attributes that are critical to the safety, efficacy, pharmacokinetics, or mechanism of action of the drug are referred to ascritical quality attributes (CQAs). The EMA has extensive experience in identifying and assessing CQAs following post-approval manufacturing changes, and has a well-established regulatory process to ensure that biosimilarity between products is maintained. Usually, only high-risk changes may require clinical data which are provided by the manufacturer in one sensitive patient population. Regulators then decide whether they want to see additional clinical trials in other indications to approve drug process modifications. To our knowledge, the EMA decided in all cases to extrapolate the clinical trial results generated in one indication to all other previously approved indications. This detailed process has been adapted to assess comparability between a biosimilar (such as the infliximab biosimilar SB2 [Flixabi®]) and its reference biological.
“There is obviously something in the art of making proteins, which we hope is quality controlled, but which needs to have checks and balances each time.” Prof Stefan Schreiber
Stepwise Assessment for the Totality of Evidence Required to Establish Biosimilarity
Regulatory approval of biosimilars requires a rigorous, stepwise assessment of the product using a combination of analytical physicochemical and biological testing, and clinical pharmacokinetic, efficacy, safety, and immunogenicity evaluations. The totality of this evidence must satisfy the requirements for biological similarity to the reference product.11
Extrapolation of Clinical Data Across Indications and Selection of a Sensitive Patient Population
According to the EMA, it may be possible to extrapolate the efficacy and safety data of a biosimilar in one clinical indication to other indications of the reference biological, provided: i) the molecular mechanism of action of the biosimilar is the same across indications and ii) there is a convincing demonstration of biosimilarity in a population that is suitable for detection of potentially clinically-relevant differences (that is, a sensitive population).7 The validity of extrapolating clinical data for biosimilars across indications has been debated by gastroenterologists. Therefore, it is important to understand the scientific rationale behind extrapolation and the validity of using clinical efficacy and safety data from one population to infer conclusions about a different patient population. Rheumatologists or dermatologists would likely feel no different to gastroenterologists.
Populations with RA and AS have been used to assess efficacy and safety outcomes of biosimilar infliximab compared with reference infliximab.12-14 RA and AS are considered to be sensitive patient populations, with well-defined outcomes (arguably better defined than in IBD) and sufficient difference in efficacy between the active treatment and placebo groups. These indications for treatment with infliximab have precise objective outcome measures for measuring disease activity, such as the ACR response criteria in RA which assesses change in swollen/tender joint count together with changes in acute phase reactant levels, patient/physician assessment, pain, and function. Furthermore, this sensitivity increases the likelihood that small differences between an active treatment and a similar treatment group will be detected.15
In addition to efficacy, it is essential to ensure that safety and immunogenicity of a biosimilar are comparable to the reference biological. Such validation needs to be assessed in a sensitive population, that is, a population that is most likely to demonstrate an immune response or an immune-related adverse event. One way of characterising immunogenicity is to measure the formation of ADAs. Data from reference infliximab clinical trials indicate that RA is the most sensitive patient population for assessing infliximab immunogenicity.16 Nevertheless, it is important to note that patients with RA typically receive infliximab at a lower dose than patients with IBD (3 mg/kg versus 5 mg/kg). They are also more often treated with concomitant methotrexate (MTX) which suppresses the formation of ADAs. However, studies comparing reference infliximab with the infliximab biosimilar CT-P13 have demonstrated that immunogenicity is similar between treatment groups in different populations (RA, AS, and CD).16,17 These findings, together with the totality of evidence generated in non-clinical comparability exercises and clinical PK studies, indicate that it is scientifically justified to extrapolate data from RA or AS populations to a population with IBD.
Analytical Testing and Biological Assays
State-of-the-art analytics are required to detect any small variations between molecules in the non-clinical analytical phase and help to ensure that any differences in CQAs fall within pre-specified limits. In the example of SB2, 46 physicochemical tests and 23 biological assays were performed to characterise the product’s primary and higher-order structures, physicochemical characteristics, and biological activities. The results of this battery of tests indicated that SB2 had consistent and uniform important product quality characteristics, allowing the EMA to conclude that the product would perform satisfactorily and uniformly in clinical practice.18 Thus, the comprehensive analytical comparability exercise demonstrated that SB2 could be considered biosimilar to reference infliximab at the quality level.18
“Developing a biosimilar is demonstrating similarity of the molecule, and the most important part is not the clinical studies, but the preclinical work.” Prof Julián Panés
Phase I Pharmacokinetic Evaluation
The EMA requires that detailed characterisation of the biosimilar in pre-clinical studies be followed by a comparative Phase I pharmacokinetic study versus its reference product.7 To show pharmacokinetic equivalence, the confidence intervals (CIs) of the test-to-reference ratio for relevant pharmacokinetic parameters between the biosimilar and the reference product must fall within a pre-specified equivalence margin, agreed with the regulatory agency.7
In the SB2 Phase I study, 159 healthy subjects were randomised to receive SB2, reference infliximab sourced in the EU, or reference infliximab sourced in the USA, all as a single dose of 5 mg/kg administered over a 120-minute intravenous infusion.19 The comparison between the EU and USA-sourced reference infliximab products was performed to demonstrate that these versions are biosimilar and to provide scientific justification for the use of EU-sourced infliximab as the only active comparator to SB2 in the Phase III study.12 In addition, the EMA suggested that the 5 mg/kg dose (recommended dose for non-RA indications) be used in the Phase I study rather than the 3 mg/kg dose (recommended dose for RA), in order to detect even minor potential pharmacokinetic differences between products.
Pharmacokinetic parameters, safety, and immunogenicity were then assessed over a 10-week period. The mean serum concentration-time profiles were superimposable between the three products, demonstrating bioequivalence. All primary endpoints i.e. the geometric, least-squares means ratios of area under the concentration-time curve from time zero to infinity (AUCinf), AUC from time zero to the last quantifiable concentration (AUClast), and maximum concentration were close to 1 for all comparisons, and the corresponding 90% CIs were completely contained within the pre-specified equivalence margin of 80–125%.19
“The molecules are the same… and they have almost completely overlaying pharmacokinetic characteristics. This is a very strong point made by this exercise.” Prof Stefan Schreiber
Phase III Clinical Efficacy Evaluation
As a final step to proving biosimilarity, the EMA requires a comparative, Phase III clinical trial between the biosimilar and the reference biological in a scientifically appropriate, sensitive patient population to demonstrate equivalent efficacy and comparable safety.7 The efficacy and safety of SB2 were compared with reference EU-sourced infliximab in a Phase III, randomised, double-blind, equivalence study performed across 11 countries in patients with moderate-to-severe RA despite treatment with MTX.12 Patients were randomised to SB2 (n=291) or reference infliximab (n=293) at a dose of 3 mg/kg, administered at Weeks 0, 2, and 6, and then every 8 weeks thereafter until Week 46. At Week 54, patients previously receiving reference infliximab were re-randomised to either receive SB2 or continue reference infliximab up to Week 70, while patients receiving SB2 continued to receive SB2 up to Week 70.20 All patients received background MTX 10–25 mg/week and folic acid 5–10 mg/week.
“We’re just making the point here that, clinically, when you have a similar molecule in the lab — physically, structurally, and functionally — you’re not supposed to find any clinical difference. But the regulators require the company to do at least one trial in a sensitive indication.” Prof Geert R. D’Haens
The primary endpoint of the study was the proportion of patients who demonstrated a 20% improvement in ACR20 at Week 30 in the per-protocol set (PPS) (Figure 1). This endpoint was met in 64.1% of patients treated with SB2 and 66.0% of patients treated with reference infliximab (treatment difference -1.88%). In the full analysis set (FAS), ACR20 response rates at Week 30 were 55.5% and 59.0% with SB2 and reference infliximab, respectively (difference -2.95%; Figure 1). The 95% CIs for the adjusted difference in ACR20 response rate fell within the pre-specified equivalence margin of ±15% in both the PPS (95% CI: -10.23% to 6.51%; primary endpoint) and the FAS (95% CI: -10.88% to 4.97%), indicating therapeutic equivalence between the products.
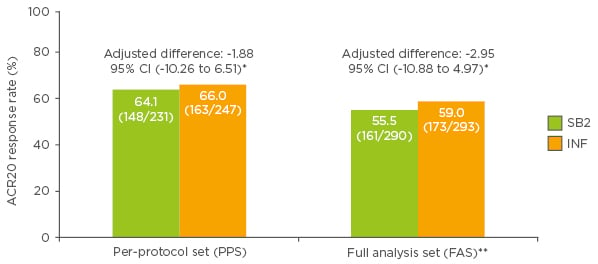
Figure 1: American College of Rheumatology 20% improvement (ACR20) response rates at Week 30 in patients treated with SB2 or reference infliximab.
*Predefined equivalence margin -15 to 15%; **patients with missing ACR20 response at Week 30 considered non-responders at Week 30.
ACR: American College of Rheumatology; CI: confidence interval; PPS: per protocol set; FAS: full analysis set; INF: reference infliximab.
Modified from Choe et al.12
The time-response curves of SB2 and reference infliximab in the FAS showed that ACR20, 50, and 70 response rates mirrored each other over the 54 weeks of the double-blind phase of the study (Biogen data on file; Figure 2). These comparable time-response rate curves support the robustness of the primary assessment of equivalence. During the extension period, the efficacy profile remained comparable up to Week 78 between patients in the reference infliximab group who transitioned to SB2 and those who remained on reference infliximab or SB2.20
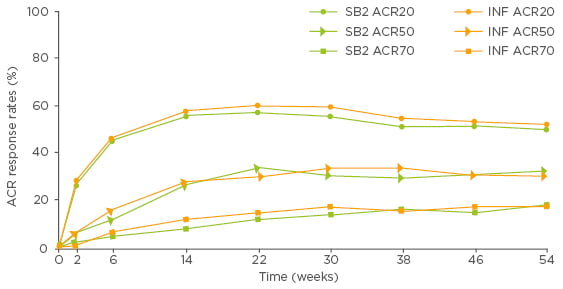
Figure 2: ACR20, ACR50, and ACR70 response rates over time up to Week 54 in patients treated with SB2 or reference infliximab (full analysis set population, Biogen data on file).
ACR: American College of Rheumatology; INF: reference infliximab.
“The superimposable time-response rate data are remarkable.” Prof Geert R. D’Haens
Whilst demonstrating between-product equivalence in terms of structural joint damage in RA was not a regulatory requirement, modified Total Sharp Score (mTSS) was assessed in both groups. The mTSS is used to assess joint damage of the hands and has been used in several trials to demonstrate differences between treatments over a 12-month period.21,22 The mean change at Week 54 from baseline in mTSS was comparable between the two treatment groups (mean change: 0.38 with SB2, 0.37 with reference infliximab).23
Phase III Clinical Safety and Immunogenicity Evaluation
In the SB2 Phase III clinical study, 62.4% of the SB2 recipients and 57.5% of the reference infliximab recipients tested positive for ADAs up to Week 54. This numerical difference in immunogenicity was not statistically significant (p=0.270).23 When excluding patients with ADA-positive results at baseline, the overall seroconverted ADA results were 54.4% in the SB2 treatment group and 48.4% in the reference infliximab group.18 As the incidence of ADAs must be evaluated in a clinical context, additional efficacy and safety analyses by ADA status were executed. These assessments demonstrated that the numerically higher incidence of ADAs in the SB2 treatment arm did not translate into any meaningful differences between SB2 and reference infliximab in terms of efficacy (data not shown) or TEAEs (Table 1).

Table 1: Incidence of treatment-emergent adverse events by anti-drug antibody status up to Week 54 in patients treated with SB2 or reference infliximab.24
*n=number of patients with TEAEs; **N=number of patients with available overall 54-week ADA assessment results. Percentages were based on N.
ADA: anti-drug antibody; TEAEs: treatment-emergent adverse events.
“This [difference in ADA levels between treatment groups] is random variation… statistical differentiation is non-existent, there is no difference.” Prof Stefan Schreiber
Following the end of the double-blind phase of the study at Week 54, the incidence of newly developed ADAs up to Week 78 was similar between treatment groups: 14.6%, 14.9%, and 14.1% in the group that transitioned from reference infliximab to SB2, the group that remained on reference infliximab, and the group that remained on SB2, respectively.20 It is notable that the rates of ADA positivity were appreciably higher both for SB2 and reference infliximab compared with historical infliximab data, suggesting that the ADA assay used here was more sensitive.
“The important thing to remember is that we are ever becoming better in finding [anti-drug] antibodies… the tests are just becoming more sensitive.” Prof Geert R. D’Haens
Transferring Clinical Data into Clinical Practice
The introduction of biosimilars will drive higher use of biological treatment, simply because there is more education and more clinical data. Furthermore, in some settings, it will free up resources to use on novel therapies and in other components of patient care.
Switching from a reference biological to a biosimilar for the same therapeutic intent is becoming an important consideration in IBD practice, particularly in terms of allowing patients to access a safe and effective treatment at a lower price. Effective switching involves monitoring immunogenicity, ensuring that there is traceability between products, and informing patients about the product that they are receiving. As similar forms of the same biological are now available and there is a high likelihood of switches between products, it is extremely important to document which product the patient has received. It may also be appropriate to use a brochure to explain to patients about the rationale and process of exchanging one biological with another biological. If a patient is concerned that the biosimilar that they have been switched to is less effective than the reference product they initially received, measuring drug levels before switching to ensure that an adequate dose is being used is recommended.
“We informed patients, monitored immunogenicity, and traced the drugs. I arranged with the hospital authorities that the savings generated would be invested back into the service through an IBD specialist nurse.” Prof Simon Travis
Data from the NOR-SWITCH trial (NCT02148640), presented at UEG Week 2016, further supported the concept of switching between a reference biological and biosimilar.25 This randomised, double-blind, non-inferiority study was performed in patients on stable, reference infliximab who were randomised to remain on the reference product or transition to an infliximab biosimilar (CT-P13). At Year 1, there were similar rates of disease worsening in both treatment groups. While patients with a range of immune-mediated diseases were enrolled in this study, it is important to note that the study was not powered to show treatment differences in each individual indication.
“[NOR-SWITCH] is further confirmation that we can switch patients safely… all patients are amenable to being switched.” Prof Julián Panés
Other transition studies are currently ongoing, which will add to the body of evidence.
“We’ve just concluded a switch trial… in 150 patients that were switched to biosimilar infliximab. The primary endpoint was pharmacokinetics — data are being generated as we speak. This, taken together with the NOR-SWITCH trial and all that we’ve seen, we’re probably becoming more compelled towards switching.” Prof Geert R. D’Haens
Conclusion
Integration of biosimilars into current IBD treatment algorithms is a reality across the EU. While biological-naïve patients are clear candidates for use of biosimilars, patients who are already on a reference biological can be considered for transition to a biosimilar after appropriate discussion with a specialist. Infliximab and other anti-TNF mAbs have an important place in the treatment of IBD. Increased use of biosimilars in patients with CD or ulcerative colitis is likely to reduce costs, increase access for eligible patients to biologic therapy, and contribute to improvements in patient outcomes.






