Abstract
Acute chest syndrome (ACS) is the most prominent cause of mortality in children with sickle cell disease. Its cause was initially not clearly understood, but there are now established concepts regarding its aetiopathogenesis. This narrative review discusses the current perspectives on sickle cell disease pathogenesis and treatment. The PubMed database was searched for articles that met the review objective. The major causative factors are pulmonary infections, pulmonary infarction, and pulmonary fat embolism from bone marrow necrosis. These factors initiate events that result in ACS, in which a vicious cycle of infarction, inflammation, and lung collapse occurs, leading to ventilation-perfusion mismatch and hypoxaemia. ACS is best managed in hospital settings because intensive care of the patient may be required. Despite its complex management, the primary treatment modalities are supportive care, transfusion therapy, and pharmacotherapy. Although the efficacy of several modalities in attenuating or preventing ACS are well established, the outcomes from instituting others are not convincing. More research is, therefore, needed to strengthen the evidence for their therapeutic efficacy.
INTRODUCTION
Acute chest syndrome (ACS) is an acute inflammatory lung injury that usually follows the vaso-occlusive crisis (VOC) seen in sickle cell disease (SCD). It is defined by the presence of a new pulmonary infiltrate on a chest radiograph, in association with fever and respiratory symptoms or signs such as cough, chest pain, hypoxaemia, wheezing, and fast breathing.1-3 It is the most prominent cause of mortality in children with SCD, occurring in >50% of patients who are admitted with VOC.4 A recent study identified young age, male sex, asthma comorbidity, and obstructive sleep apnoea as significant risk factors for ACS.5 Additionally, young age and a history of asthma are associated with recurrence of ACS.6 Risk factors and symptoms of both asthma flare-ups and ACS are remarkably similar.7 Nevertheless, a multicentre prospective study observed that older patients (aged ≥20 years) experienced a more severe clinical course than their younger counterparts.8
Although the pathogenesis of the syndrome was thought to be mostly idiopathic, there are now established concepts about its aetiopathogenesis. For instance, a recent narrative review indicated that the aetiologic factors consist of pulmonary infections, pulmonary infarction, and pulmonary fat embolism, which originate from bone marrow necrosis.9 The underlying pathogenic mechanism involves a hypoxia-induced increase in intercellular adhesion of sickled red blood cells and their increased adhesiveness to the endothelial lining of the pulmonary vasculature. This is facilitated by the interaction between erythrocytic very late activation antigen-4 and endothelial vascular cell adhesion molecule-1 (VCAM-1).10 The resultant vaso-occlusion initiates a vicious cycle of pulmonary infarction, inflammation, and lung collapse. The clinical deterioration in ACS is specifically caused by the initial hypoxia-induced upregulation of pulmonary endothelial VCAM-1, which is not neutralised by the reactive formation of cytoprotective mediators such as nitric oxide (NO); this leads to intrapulmonary adhesion.10
A clear understanding of the causes and pathogenesis of ACS and applying appropriate therapeutic interventions may prevent fatal outcomes. The present narrative review discusses the current perspectives on the pathogenesis and treatment of this syndrome. It presents a discourse on the effective modalities for managing ACS in children with SCD.
LITERATURE SEARCH: STRATEGY AND OUTCOME
A search of the PubMed database using the descriptors “acute chest syndrome” and “sickle cell disease” yielded 931 published articles. Addition of search terms such as “children” and “pathogenesis and treatment” scaled down the number of articles to 539 and 288, respectively. After exclusion of duplicates, case reports, items unrelated to the review objective, and letters to the editor, 102 original and review articles (published in the English language) were selected. Additionally, the items that met the inclusion criteria for review consisted of those published from the previous four decades to the current date. Relevant information was extracted from these papers for the present narrative review.
RISK FACTORS FOR ACUTE CHEST SYNDROME
Several modifiable and nonmodifiable risk factors of ACS have been identified from numerous studies. These risk factors include young age,5,7,11 SCD genotypes such as HbSS or HbS-ß0 thalassaemia,11,12 asthma comorbidity,6,13-17 low proportion of fetal haemoglobin,11 high steady-state haemoglobin concentration,11 high steady-state leukocyte count,11,13,18 primary or secondary tobacco smoke exposure,19-21 more than three episodes of VOC in the previous year,13 and recent surgical intervention.22
Some of these factors can predict the occurrence of ACS based on the following possible mechanisms. Firstly, high asthma prevalence rates in children with SCD14,23 underscore the vital role of asthma in the development of ACS. Several studies have noted a direct relationship between asthma flare-ups and episodes of ACS at a young age.16,17,24-26 It does appear that children with SCD and comorbid asthma are very likely to develop respiratory symptoms during a VOC.25 The suggested mechanisms for the predisposition to ACS include a damaged vasculature, bronchospasm-induced hypoxia leading to increased red blood cell sickling and subsequent vaso-occlusion, as well as the inflammation arising from disease synergy. These mechanisms contribute to and accentuate the vicious cycle of pulmonary infarction, inflammation, and lung collapse, which characterise ACS. Secondly, the high steady-state haemoglobin concentration and leukocyte count associated with ACS risk are thought to be contributory factors through their alteration of blood rheology. The increased blood viscosity promotes vaso-occlusive events. However, after development of VOC, acute haemolysis may contribute to the evolution of pulmonary injury through elevated amounts of free circulating haem. This product of haemolysis contributes to the development of ACS in paediatric patients with SCD.27 Its role in the pathogenesis of the syndrome has been highlighted subsequently in this review. Finally, ACS triggered by secondary exposure to tobacco smoke is explained by mechanisms such as lower airway obstruction in children with SCD, as well as increased expression of VCAM-1 with airway inflammatory response.28
Notably, there are clear differences between the clinical presentation of ACS seen in children and adults with SCD. Whereas ACS in children usually presents at admission as a mild disease simulating community-acquired or viral pneumonia, the onset of the syndrome in adults is usually 1–3 days after hospitalisation for a severe VOC or bone pain crisis. Additionally, ACS in adults often runs a rapidly progressive course which may culminate in respiratory failure and is thus associated with high mortality rates. These differences may be attributable to the high prevalence of fat embolisation in adults and community-acquired or viral pneumonia in children.8
PATHOGENESIS OF ACUTE CHEST SYNDROME
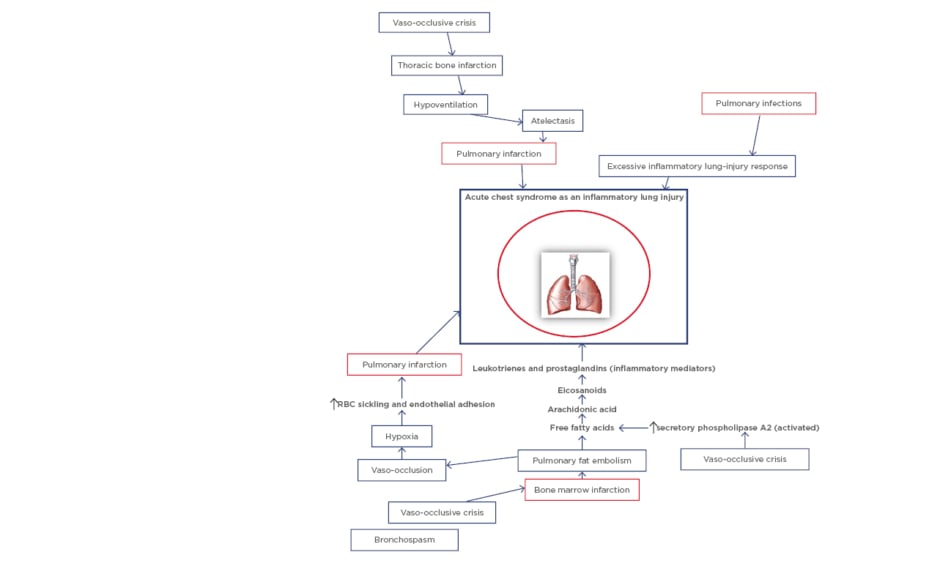
An overview of the major pathogenic pathways involved in SCD is important in understanding the pathogenesis of ACS. Sickled red blood cells that arise from hypoxia-induced formation of sickle haemoglobin (HbS) polymers follow either the haemolytic or vaso-occlusive pathways. In the former, haemolysis releases free circulating haem which contributes to systemic endothelial dysfunction (haemolysis-related vasculopathy). On the other hand, sickled red blood cells can sequentially lead to vascular stasis, vaso-occlusion, and infarction (viscosity-vaso-occlusive phenomenon). The subsequent ischaemia-reperfusion results in systemic inflammation which also contributes to systemic endothelial dysfunction, characterised by increased adhesion molecules, reduced NO bioavailability, and increased vascular permeability. The systemic endothelial dysfunction is followed by increased red-cell and white-cell adhesion to vascular endothelium in target organs. For instance, two different phenotypes on pathogenesis have thus been proposed for sickle-cell nephropathy, namely ‘haemolysis-endothelial dysfunction’ phenotype and ‘viscosity-vaso-occlusive’ phenotype.29 These phenotype models also equally explain some of the pathogenic pathways involved in SCD-related pulmonary complications. ACS, as an example of acute pulmonary injury, is initiated by multiple causative factors. The pathogenesis of the syndrome revolves around these major factors comprising pulmonary infections, pulmonary infarction, and pulmonary fat embolism (Figure 1).
Figure 1: Schematic diagram of the pathogenesis of acute chest syndrome.
RBC: Red blood cell.
Specific bacterial and viral pathogens have been identified as common infectious agents.30 These include atypical bacteria such as Chlamydia pneumoniae and Mycoplasma pneumoniae, Staphylococcus aureus, Streptococcus pneumoniae, and respiratory syncytial and influenza viruses. The infectious agents are believed to trigger ACS through excessive inflammatory response in the lungs, which arises from the systemic endothelial dysfunction.10,31 Secondly, another pathway leading to ACS involves increased endothelial adhesion by sickled red cells and subsequent vaso-occlusion and pulmonary infarction: the viscosity-vaso-occlusion phenotype. The pulmonary infarction results in ventilation-perfusion (VQ) mismatch and worsening of hypoxaemia. Finally, pulmonary fat embolism triggers episodes of ACS through the pathogenic trajectory in which, at the onset, the VOC leads to bone marrow infarction and necrosis. This complication then deposits fat droplets into the circulation. Fat embolisation into the pulmonary vasculature initiates a metabolic cascade in which activated secretory phospholipase A2 cleaves fatty acids from phospholipids, releasing arachidonic acid. Subsequently, arachidonic acid is metabolised into eicosanoids which consist of the inflammatory mediators leukotrienes and prostaglandins; these molecules mediate alveolar inflammation and injury to the pulmonary vascular endothelium.32 Similarly, haem released from haemolysis contributes to the pulmonary vasculopathy (haemolysis-endothelial dysfunction phenotype), and initiates the development of ACS. In summary, ACS is characterised by a vicious cycle of pulmonary infarction, inflammation, and lung collapse, which is associated with VQ mismatch and hypoxaemia.33 Furthermore, a study suggests that episodes of ACS render children with SCD more susceptible to increased lower airway obstruction.34 This finding lends credence to the high case fatality rates associated with ACS in these children.
TREATMENT MODALITIES FOR ACUTE CHEST SYNDROME
ACS is best managed in hospital settings because intensive care of the patient may be required. The bedrock of treatment is supportive care. Recently, an expert-panel report from the National Heart, Lung, and Blood Institute (NHLBI) released evidence-based guidelines which have led to the effective treatment of SCD-related complications, including ACS.35 Although the management of ACS is considered complex, the primary treatment modalities comprise supportive care, transfusion therapy, and pharmacotherapy (Table 1).
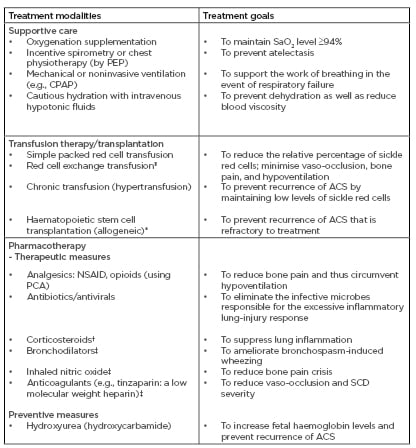
Table 1: A summary of treatment modalities in acute chest syndrome.
ACS: acute chest syndrome; CPAP: continuous positive airway pressure; NSAID: non-steroidal anti-inflammatory drugs; PCA: patient-controlled analgesia; PEP: positive expiratory pressure; SaO2: arterial oxygen saturation; SCD: sickle cell disease.
†: use still controversial; ‡: further research needed to validate its use; *: proven effective in children (modality of SCD cure); §: automated red cell exchange using a machine (erythrocytapheresis) or by manual red cell exchange (phlebotomy and transfusion).
The essential components of supportive care consist of sufficient oxygenation and ventilation, prevention of lung collapse, and cautious hydration. The goal of supplemental oxygen is to maintain the arterial oxygen saturation (SaO2) level at ≥94%. Thus, ACS patients with SaO2 values <94% or fast breathing require prompt oxygenation. When compared to pulse oximetry, which determines peripheral oxygen saturation level, arterial blood gas determination of SaO2 is an ideal and accurate technique for measuring hypoxaemia in SCD patients who have abnormal haemoglobin-dissociation curves.36 This pathophysiologic characteristic poses a challenge in resource-limited settings where pulse oximetry is more likely to be the available and accessible tool to measure hypoxaemia. Furthermore, inadequate monitoring and correction of hypoxaemia will worsen the VQ mismatch and contribute to fatal outcomes. Occasionally, SCD patients with chronic hypoxaemia may preclude the adoption of 94% as the limiting SaO2 value for oxygenation. In this instance, a much lower value deviating from the baseline steady-state SaO2 may be used to determine therapeutic intervention.
Signs of respiratory failure such as deteriorating hypoxaemia, severe breathing difficulty, and respiratory acidosis, may warrant supporting the work of breathing using noninvasive or mechanical ventilation. In fact, children with respiratory failure or disease progression who require ventilation support are best monitored and treated in paediatric intensive care units.2 Despite the previous report indicating scant evidence for the use of noninvasive ventilation (continuous positive airway pressure) in children with ACS,37 a more recent study suggests that its early use with blood transfusion was well tolerated in affected children.38 Nevertheless, the role of noninvasive ventilation in changing patient outcomes has also been investigated in adult patients with ACS. A prospective randomised study reported improvements in respiratory outcome measures such as respiratory rate and gas exchange in these adult patients, albeit transient, but failed to document any change in blood transfusion rates: the intervention being associated with patient discomfort.39
A related component of supportive care is the prevention of lung collapse, which helps to circumvent the occurrence of hypoxaemia and VQ mismatch. Recommended methods of preventing lung collapse include incentive spirometry in wakeful state40 and chest physiotherapy by positive expiratory pressure for younger patients who are either not able to comply with incentive spirometry or are breathing-constrained by thoracic wall pain.41 In fact, both incentive spirometry and positive expiratory pressure resulted in the same outcome of preventing progression to ACS in a study of hospitalised children with SCD presenting with chest pain.42 Furthermore, other authors have reported that incentive spirometry for children with SCD admitted without respiratory symptoms led to a decline in the incidence of ACS, especially for the cohort presenting with back pain.40
Hydration is also an essential aspect of supportive care. However, intravenous hypotonic fluids should be administered with caution because patients with ACS are susceptible to developing pulmonary oedema.43 To circumvent pulmonary oedema, it is vital to deliver 75% of daily maintenance fluid and avoid intravenous bolus fluid.9
Concerning transfusion therapy, both simple and exchange blood transfusions are recommended in the treatment of ACS. Although an updated Cochrane review did not find enough evidence from randomised control trials to either support or refute the use of blood transfusion, the authors suggested that transfusion therapy should be individualised and experience driven.44 According to the NHLBI expert-panel report, simple packed red blood cell transfusion (10 mL/kg) in ACS should aim to raise the haemoglobin level to 9–11 g/dL, or decrease the HbS level to >30%.35 Compared to exchange blood transfusions, simple blood transfusions may not be able to reduce the HbS to this recommended target level without causing volume overload. Exchange blood transfusion therapy will perhaps be more beneficial in achieving this treatment goal because it increases the percentage of normal haemoglobin. At the same time, it reduces the percentage of HbS and the risk of iron overload from increased volume of transfused blood, and maintains the haematocrit levels at safe limits. It is therefore mostly used in patients who are not very anaemic.
Nevertheless, automated red cell exchange (RCE) transfusion or erythrocytapheresis is the recommended transfusion therapy for SCD patients who have severe and worsening ACS.45 Similar to the effect of simple red cell transfusion, a double-volume RCE transfusion reduces the percentage of HbS to <20% and augments vascular perfusion.46 This finding confirms a previous observation that both forms of blood transfusion result in the same outcomes in patients with ACS.22 More importantly, double-volume RCE transfusion effectively, but transiently, reduces plasma inflammatory mediators such as white blood cells, absolute neutrophil count, platelets, and soluble VCAM-1 level.47 This action may help mitigate the pulmonary vaso-occlusive sequelae that characterise ACS. Despite these advantages, the superiority of RCE transfusion over simple red cell transfusion has not been well established in clinical trials; an area that requires further investigation with appropriate study designs. Chronic blood transfusion, or hypertransfusion, is another type of transfusion therapy which is indicated in SCD patients with frequent ACS because it helps to prevent ACS recurrence by maintaining low levels of sickled red cells. However, it is potentially associated with the complication of iron overload. Finally, haematopoietic stem cell transplantation is advocated for SCD patients with eligibility criteria that consist of age <17 years, availability of HLA-matched sibling donor, and several complications which include severe and recurrent ACS.48 Haematopoietic stem cell transplantation can prevent the recurrence of ACS, especially in cases refractory to pharmacologic agents such as hydroxyurea.
ACS is amenable to treatment with various pharmacologic agents. Firstly, antibiotics occupy a prominent place, given the role of pulmonary infections in its aetiopathogenesis. Empirical antibiotic therapy is based on the already documented epidemiologic patterns.30 Thus, a regimen that comprises macrolides and third-generation cephalosporin with or without vancomycin is recommended as an ideal antibiotic therapy. However, with overuse of antibiotics and the consequent emergence of multidrug resistant bacteria, it may eventually become auspicious to institute routine blood cultures of pathogens to establish antibiotic-sensitivity patterns. A 10-day course of antibiotic therapy is considered optimal, although there are currently no guidelines on the maximum duration of therapy.2 Administering antiviral agents has also been strongly advocated when seasonal influenza is under consideration, given the fact that children with ACS are more likely to have a viral aetiology than a bacterial aetiology.
Secondly, analgesic therapy is relevant in ACS patients because chest wall pain may be associated with thoracic splinting and hypoventilation, and is directly related to the evolution of the syndrome.49 The current practice on pain management involves the use of a ‘step-ladder’ approach in which mild-to-moderate pain is treated with nonsteroidal anti-inflammatory drugs, while severe pain is managed with opioid analgesics, especially morphine.50 However, in using the latter, the risk-benefit consideration is essential as the aim of treatment remains to achieve analgesia to obviate hypoventilation, while simultaneously avoiding opioid-related respiratory depression.2 Thus, to achieve proper dosing of opioids to minimise these side-effects, patient-controlled analgesia could be used.51 Parenteral opioids are administered through a microprocessor-controlled infusion pump which is used by patients once pain sensation occurs. Patient-controlled analgesia works on the basic principle of the patient’s ability to evaluate pain intensity in VOC as well as analgesic potency. At the onset of pain, the patient activates the dosing system and receives a doctor-programmed analgesic dose, which is followed by activation of a protection system to prevent overdose.
With comorbid asthma and bronchospasm as a contributory risk factor for ACS, treatment with inhaled bronchodilators is advised, more so with the high incidence of lower airway obstruction and wheezing reported in ACS patients.34 However, the evidence for their efficacy remains weak according to a recent Cochrane review.52 Another inhalational pharmacologic agent adjudged as a potential medication in VOC is NO. On the contrary, a randomised control trial in hospitalised SCD patients with VOC did not document any significant difference in the resolution of the crisis and the episodes of ACS between NO and placebo groups.53 Regarding the use of corticosteroids in ACS, which still remains controversial, reports show that when administered in a low dose the drug could improve disease morbidity54 and reduce the need for blood transfusion.55 Nevertheless, documented adverse events such as haemorrhagic cerebrovascular accident56 and recurrence of VOC after drug withdrawal57,58 represent drawbacks to its regular use in ACS. Recently, anticoagulant therapy has also been considered as a modality in managing ACS. Specifically, tinzaparin (low-molecular-weight heparin) is known to reduce the duration and severity of pain associated with VOC by ameliorating vaso-occlusion.59
Hydroxyurea (hydroxycarbamide) is another pharmacologic agent that has been approved and used over the years as a disease modifier. It induces production of fetal haemoglobin with a concomitant decrease in the intracellular concentration of HbS, which affects the polymerisation of deoxygenated HbS. It also reduces the white cell count and the expression of cell adhesion molecules that contribute to vaso-occlusion and may also serve as a NO donor.60,61 These actions underpin its role in the reduction of recurrence of ACS in children. Although there were initial concerns regarding its toxicity from long-term exposure, there is now reliable evidence supporting its use even in patients as young as 9 months of age, given its ability to reduce the frequency of VOC and ACS with minimal or absent side effects.62
CONCLUSION
ACS is associated with high mortality rates in children with SCD. Its aetiopathogenesis is predicated upon pulmonary infections, pulmonary infarction, and pulmonary fat embolism. The syndrome is characterised by a vicious cycle of pulmonary infarction, inflammation, and lung collapse, which is associated with VQ mismatch and hypoxaemia. Effective management thus requires a meticulous approach by the clinician to institute the primary treatment modalities comprising supportive intensive care, transfusion therapy, and appropriate pharmacotherapy. Whereas the efficacy of some of these modalities in attenuating or preventing ACS are well established, outcomes from instituting others are not yet convincing. For instance, more research is still needed to establish the superiority of exchange blood transfusion over simple blood transfusion, as well as the effectiveness of corticosteroids and bronchodilators in the management of the syndrome in children. Anticoagulant therapy using low-molecular-weight heparin (tinzaparin) also merits further attention and investigation.





