Abstract
Amyloidoma is a rare complication of plasmacytoma, that can involve the spine and present with compressive neurological symptoms. It is usually a diagnosis of exclusion, and is difficult to differentiate from other plasma cell disorders on imaging. Definite diagnosis requires a tissue biopsy. The treatment requires a multidisciplinary approach, with input from haematology, neurology, neurosurgery, and radiology for the optimum course of action, depending on the patient’s comorbidities and performance status. The authors hereby present a case of solitary sacral amyloidoma in a 52-year-old African American female. Only three other cases of solitary sacral amyloidoma have been reported in the literature.
Key Points
1. Plasmacytoma of the spine can present with residual amyloidoma after treatment.2. The diagnosis of amyloidoma requires biopsy to exclude the mimics.
3. The treatment of spinal amyloidoma is challenging due to its close vicinity to the spinal cord, but early neurosurgical intervention has been shown to improve outcomes.
INTRODUCTION
Amyloidosis results from a misconfiguration of normal protein into β-plated linear sheets, leading to the formation and deposition of insoluble fibrils.1 These fibrils can sometimes form a localised tumour-like mass called amyloidoma at specific organ sites, even in the absence of systemic amyloidosis.2 Spinal amyloidoma can present with symptoms of nerve compression and radiculopathy, depending upon the site of involvement.3 Amyloidoma of the bone can sometimes be difficult to differentiate from other pathologies that commonly cause lytic lesions, like multiple myeloma (MM), plasmacytoma, and metastatic diseases.4 Therefore, the definitive diagnosis requires a tissue biopsy and Congo red staining, which produces an apple-green birefringence under polarised light.
PATIENT INFORMATION
A 52-year-old African American female with a past medical history of IgG lambda MM and sacral plasmacytoma presented with worsening back pain, bowel and bladder incontinence, bilateral lower extremity weakness, and saddle anaesthesia concerning for cauda equina syndrome. She had been diagnosed with IgG lambda MM with paraprotein level of 0.2 g/dL 10 months previously, and had received radiation and bortezomib, lenalidomide, and dexamethasone, with subsequent normalisation of the light chain ratio, and complete disappearance of paraprotein. She had declined autologous stem cell transplant. The MRI of lumbosacral spine carried out at the time of plasmacytoma diagnosis showed an expansile and enhancing soft tissue mass overlying the right sacral ala, while the biopsy at that time showed presence of plasma cell neoplasm without any Congo red staining.
CLINICAL FINDINGS AND DIAGNOSTIC ASSESSMENT
On presentation, initial lab work revealed normal levels of haemoglobin, creatinine, calcium, and serum protein. MRI of the lumbar spine revealed an irregular expansile enhancing soft tissue mass along the right hemisacrum from the S1 to S4–S5 level, encompassing the right neural foramina of S2, S3, and S4 (Figure 1). These findings were identical to the CT scan finding carried out 2 months previously. Furthermore, serum immunoglobulin, serum protein electrophoresis, urine protein electrophoresis, and free light chain ratio were also normal (Table 1). A PET scan was carried out 1 month prior to the presentation, and did not show FDG uptake or evidence of metastasis in the area. With these findings, the suspicion of MM, plasmacytoma recurrence, and malignancies were low, leaving a suspicion for a sacral amyloidoma (SaA) as the diagnosis of exclusion. CT-guided biopsy was performed, which revealed a proteinaceous material showing apple-green birefringence under polarised light following Congo red stain, without evidence of plasma cell neoplasm, consistent with SaA (Figure 2). This was unlike the biopsy which occurred 10 months prior, at the time of initial diagnosis, which showed sheets of mature-appearing plasma cells. Furthermore, a colonoscopy with biopsy and cardiac MRI were also done, which ruled out systemic amyloidosis.
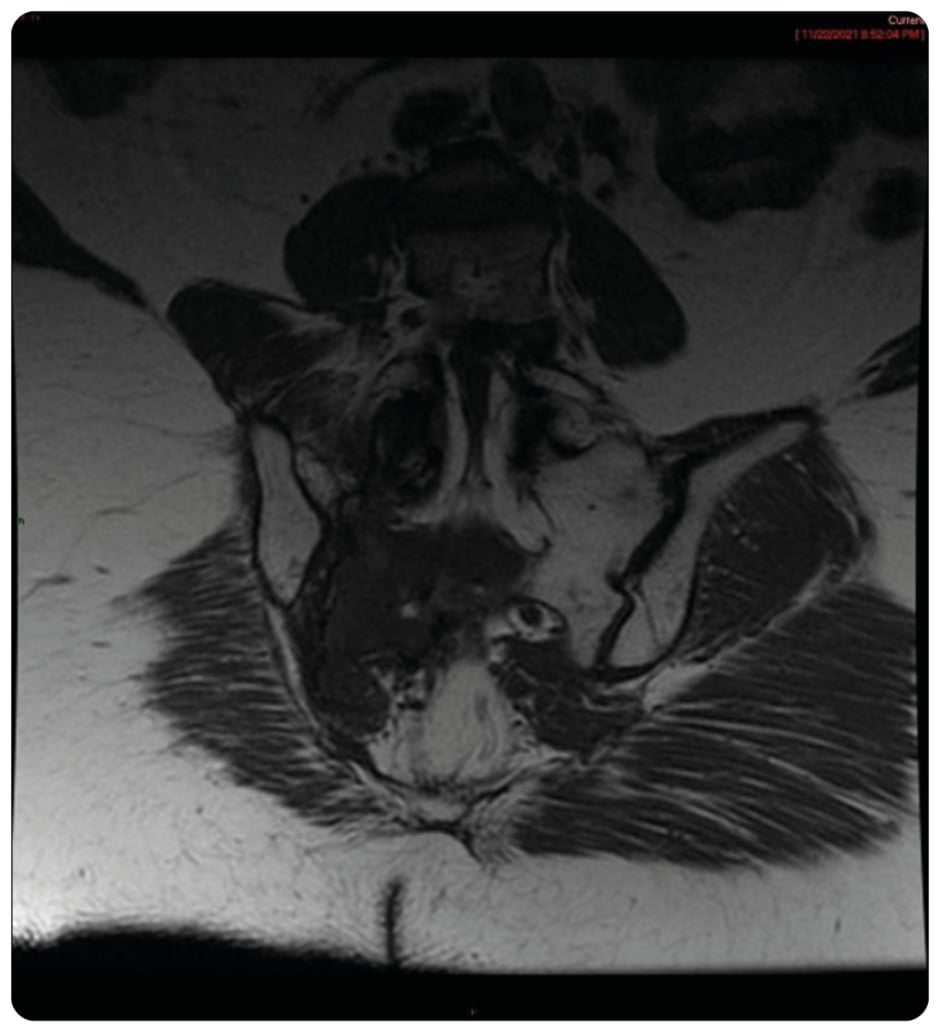
Figure 1: Coronal T1 showing large inferior T1 mass in right sacrum crossing midline.
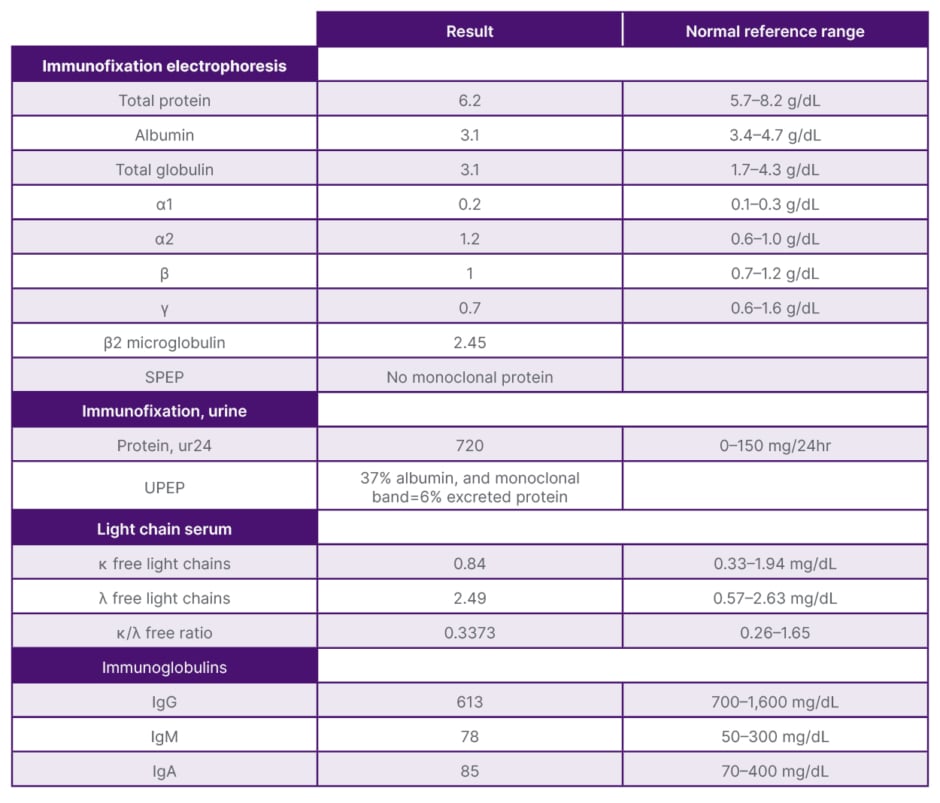
Table 1: Pertinent laboratory findings.
SPEP: serum protein electrophoresis; UPEP: urine protein electrophoresis.
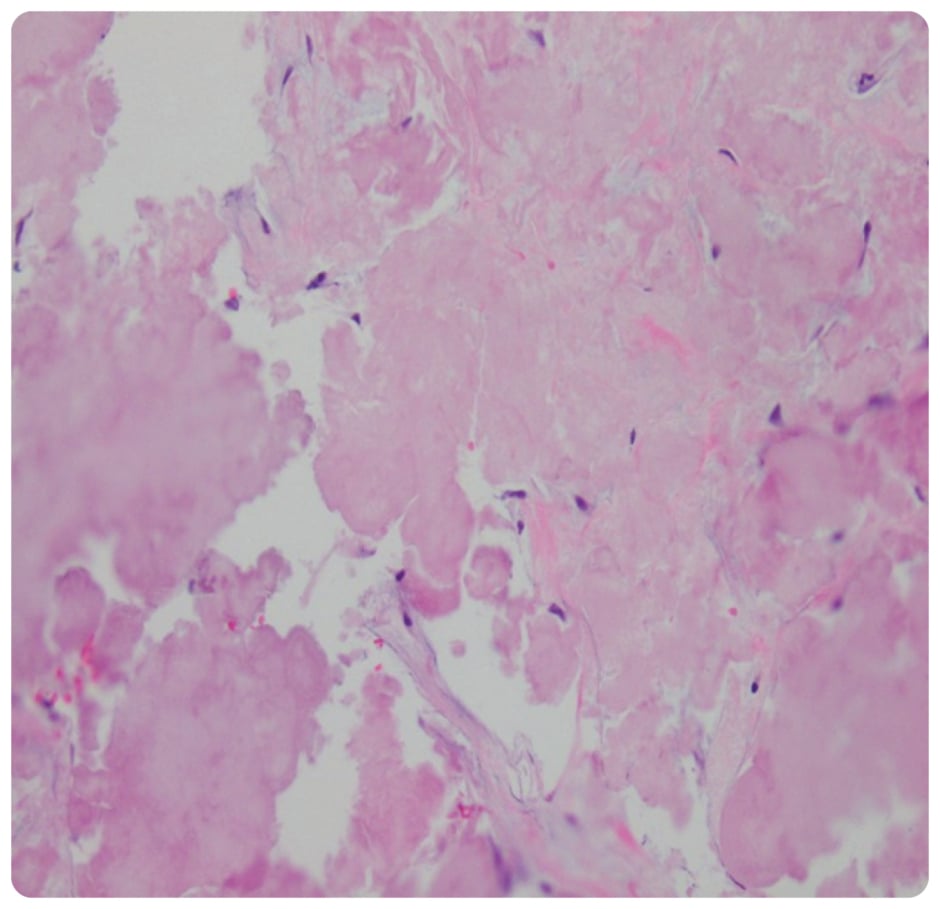
Figure 2: Biopsy showing proteinaceous material with no evidence of neoplastic plasma cells.
THERAPEUTIC INTERVENTION
The patient was started on dexamethasone, which helped to improve the back pain and incontinence within days. Hospital stay was further complicated by methicillin-sensitive staphylococcal bacteraemia, acute anaemia (haemoglobin <7 g/dL), and sacral fracture. The patient was managed conservatively, considering she had improvement of neurological symptoms with steroids; the absence of a fat pad between the spinal cord; and the large size of the mass, which placed her at a high risk of surgical and long-term neurological complications. In addition, bacteraemia made her a high-risk surgical candidate. She received dexamethasone 40 mg daily for 4 days. She was discharged with a spinal brace for outpatient follow-up.
FOLLOW-UP AND CLINICAL OUTCOMES
A repeat CT scan 2 months later revealed the mass was stable, and MM remained in remission without biochemical progression.
DISCUSSION
Amyloidoma is an aggregate of misfolded protein localised in an organ. It commonly involves the bladder, larynx, tonsils, skin, and lungs.5 Solitary spinal amyloidoma (SSA) is rare, with sacral spinal amyloidoma being exceptionally uncommon. In a review of literature, there were a total of 38 reported cases of SSA, out of which 18 were thoracic, 13 were cervical, four were lumbar, and only three were sacral.3,4,6-9 Like plasmocytomas, amyloidomas are usually found in the elderly, with 74% of cases being diagnosed between 60–80 years of age.3 The patient, however, had an usually early presentation at the age of 52, which may be due to her history of MM and sacral plasmacytoma, and may represent disease continuum. The exact mechanism or factors leading to transformation of the spinal lesion from plasmacytoma to amyloidoma is unknown, and needs further research. As amyloidoma is an expansile mass, the symptoms are generally progressive and are related to the site and neurological involvement.3-9 The most common presenting symptoms in patients with SSA, as expected, are either pain or neurologic compromise, with decreased muscular strength (74.29%), and axial back pain (65.70%) reported. However, only 17% of cases present with acute symptoms.3 The patient had chronic back pain, likely due to the evolving mass, but presented with cauda equina syndrome when the mass encroached the spinal cord.
For every patient with cauda equina syndrome, initial imaging starts with an emergent MRI/CT myelography of the spine.10 An SSA generally appears as a hypointense lesion on T1- and T2-weighted images.3 CT imaging generally reveals lytic bone lesions with cortical destruction, and possibly pathological fractures, further raising diagnostic challenges, as the findings mimic MM and other primary neoplasms, like plasmacytoma, chondrosarcoma, or secondary metastatic lesions.11 A biopsy is required to distinguish between SSA and other mentioned differentials. An apple-green birefringence when stained with Congo red stain visualised under polarised light, without evidence of plasma cell neoplasm, is classically seen.3-5 Moreover, it should be noted that excluding systemic involvement is of utmost importance in such cases, to rule out systemic amyloidosis.
The treatment of the SSA is challenging, due to its close vicinity to the spinal cord, but early neurosurgical intervention has shown to improve outcomes. Based on the review by Pinherio et al.,3 out of 29 patients who underwent surgical resection for a spinal amyloidoma, 24 patients (82.76%) had post-operative neurological improvement. Out of the three cases of SaA, two underwent surgery, and both showed neurological improvement. Another case of SaA reported by Griffin et al.9 had no neurological symptoms, and therefore did not undergo surgical intervention. Based on few reports, surgery seems to be a reasonable approach in a symptomatic patient like this one. However, in this case, the authors elected for conservative management due to high surgical risk, and improvements in neurological symptoms with steroids.
CONCLUSION
Although cases of distant amyloidoma arising secondary to plasmacytoma have been reported, as per the authors’ knowledge, this is the first case report of plasmacytoma of the spine with residual amyloidoma after treatment. In cases of SaA presenting as cauda equina syndrome, it is imperative to initiate steroid therapy, and seek a neurosurgery consult for consideration of emergent decompression surgery.