Abstract
With the increasing burden of liver cirrhosis, the most advanced stage of hepatic fibrosis, there is a need to better understand the pathological processes and mechanisms to target specific treatments to reverse or cease fibrosis progression. Antiviral therapy for hepatitis B and C has effectively treated underlying causes of chronic liver disease and has induced fibrosis reversal in some; however, this has not been targeted for the majority of aetiologies for cirrhosis including alcohol or nonalcoholic steatohepatitis. Fibrosis, characterised by the accumulation of extracellular matrix proteins, is caused by chronic injury from toxic, infectious, or metabolic causes. The primary event of fibrogenesis is increased matrix production and scar formation mediated by the hepatic stellate cell, which is the principal cell type involved. Experimental models using rodent and human cell lines of liver injury have assisted in better understanding of fibrogenesis, especially in recognising the role of procoagulant factors. This has led to interventional studies using anticoagulants in animal models with reversal of fibrosis as the primary endpoint. Though these trials have been encouraging, no antifibrotic therapies are currently licenced for human use. This literature review discusses current knowledge in the pathophysiology of hepatic fibrosis, including characteristics of the extracellular matrix, signalling pathways, and hepatic stellate cells. Current types of experimental models used to induce fibrosis, as well as up-to-date anticoagulant therapies and agents targeting the hepatic stellate cell that have been trialled in animal and human studies with antifibrotic properties, are also reviewed.
INTRODUCTION
The burden of chronic liver disease continues to grow, with 0.1% of the European population affected by cirrhosis, the most advanced stage of hepatic fibrosis.1 Although the aetiology of the disease varies between countries, fibrogenesis is the common pathological mechanism that causes cirrhosis. Fibrosis occurs following chronic liver injury from a range of insults including toxins (alcohol), infections (hepatitis B [HBV] and C viruses [HCV]), and metabolic disease (nonalcoholic fatty liver disease). Such insults drive inflammation, resulting in increased synthesis and altered deposition of extracellular matrix (ECM) components, and impaired regeneration and wound healing responses.2 This is a complex, dynamic process, involving recruitment and activation of platelets, inflammatory cells, hepatic stellate cells (HSC), and other ECM-producing cells including portal fibroblasts, hepatocytes, cholangiocytes, and bone marrow-derived cells.3 The end result, cirrhosis, is defined by profound distortion of hepatic microarchitecture, ultimately resulting in the development of portal hypertension.4 Histologically, cirrhosis is characterised by regenerative nodules of liver parenchyma separated by, and encapsulated in, fibrotic septa, with a clinical consequence of increased mortality, morbidity, complications of portal hypertension, and diminished quality of life.2,5 The presence of hepatic fibrosis is a key predictor of prognosis in chronic liver disease, independent of aetiology.6Generally this process evolves over decades (usually 20–40 years), but it can be rapidly progressive, as seen in children affected by biliary atresia, drug-induced liver injury, HCV co-infection with HIV, or HCV infection post liver transplantation.7
Management of chronic liver disease has largely focussed on aetiology-specific treatments; however, significant progress has been made in understanding the pathophysiology of fibrosis, which has identified targets for potential antifibrotic agents to either halt progression or reverse fibrosis.
A search of the existing literature up to June 2019 was conducted using electronic databases PubMed, Medline, and the Cochrane library, as well as relevant guidelines, to present this literature review, an overview of the current understanding of the pathogenesis of hepatic fibrosis, in vitroandin vivomodels used in exploring pathogenesis, and an update on proposed therapies.
PATHOPHYSIOLOGY
Extracellular Matrix
The ECM is a dynamic structural component of the normal liver.8 It contains macromolecules that provide the scaffolding of the liver and act as transducers of extracellular signals. In normal liver tissue the ECM is composed of collagens, glycoproteins, fibronectin, laminin, tenascin, von Willebrand factor, and proteoglycans.9It is a component of Glisson’s capsule, portal tracts, central veins, and the subendothelial space of Disse.10Various cellular sources of the ECM have been identified but the main source is the HSC. When liver injury is not severe, neighbouring hepatocytes regenerate and replace apoptotic and necrotic cells. However, in chronic insult, the mechanism contributing to fibrosis, this process fails and ECM proteins take on the role of hepatocytes.11 The quantity and quality of the ECM changes, increasing up to 8-fold compared to a normal liver. There is significant increase in collagen content and proteoglycans, resulting in a higher density interstitial type matrix.12The ECM composition also transforms, from one predominantly made up of Type IV and VI collagen, glycoproteins, proteoglycans, and laminin, to a matrix consisting of Type I and III collagen and fibronectin.10 These adaptations alter the local microenvironment leading to subsequent functional and physical restrictions on plasma flow between sinusoids and hepatocytes, causing impaired hepatic function.9
Signalling in Fibrosis
Fibrogenesis is a complex mechanism involving an array of cellular and extracellular signalling. Cytokine, chemokine, adipokine, neuroendocrine, angiogenic, and nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) signalling have all been found to play important functions.13The release of cytokines in the context of fibrogenesis is controlled in part by activation of metalloproteinases. TGF-β1, platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), TNF-α, and vascular endothelial growth factor (VEGF) are all involved in key mechanisms of fibrosis.9,14TGF-αis particularly important in mediating stellate cell activation to myofibroblasts and stellate cell collagen production.14VEGF and PDGF control angiogenesis, which contributes to ECM production, portal hypertension, and regeneration postinjury.15During acute or chronic liver injury, platelets are the first cells recruited to the injury site. They form aggregates in damaged vasculature by converting fibrinogen into fibrin and platelet α-granules rich in PDGF, TGF-α, and VEGF.16
Chemokines, CCL2 (produced by Kupffer cells and HSC), and CCL5 are best recognised in fibrosis.17CCL2 promotes HSC activation, facilitating macrophage and monocyte recruitment into the liver. Inhibition of CCL2, or its receptor CCR2, is associated with reduced fibrosis in experimental models.18CCL5 and its receptors, CCR1 and CCR5, are associated with fibrogenesis promotion.19Other signalling molecules involved in fibrogenesis include the adipokines, leptin, and adiponectin, synthesised by HSC, as well as cannabinoids and their respective receptors. Cannabinoid receptor 1 is a profibrotic mediator; however, cannabinoid receptor 2 has antifibrotic, but proinflammatory, properties.13
Reactive oxygen species mediate fibrogenesis by stimulating profibrogenic mediator release from Kupffer cells and activating HSC. Emerging evidence suggests that NADPH also plays a role in this process.20
Hepatic Stellate Cells
The HSC is the principal cell type involved in fibrogenesis21and its activation is the common pathway leading to fibrosis. Developing a clear understanding of factors that stimulate or inhibit activation of HSC has helped guide development of antifibrotic therapies. HSC lie in a quiescent state in the subendothelial space of Disse and comprise 15% of liver cells.8In the normal liver, they are the primary storage site for retinoids and are derived from neural crest tissue because of expression of neural crest markers, including glial fibrillary acidic protein and nestin.22These cells are activated by proinflammatory cytokines and protease activated receptor-1 ligation, resulting in a myofibroblastic phenotype which is proliferative, fibrogenic, and contractile.
Activation of HSC proceeds through an initiation and perpetuation phase.13
Initiation refers to early changes that occur in gene expression and phenotype, initiated by mediators that render quiescent HSC responsive to other stimuli. Characteristic to this phase is the production of a fibrogenic, contractile phenotype with induction of PDGF receptors.22Initiation is stimulated by oxidant stress signals, apoptotic hepatocytes, lipopolysaccharides, and paracrine stimuli from neighbouring cells including cholangiocytes, Kupffer cells, injured sinusoidal endothelial cells, or other stellate cells.13Initiation activates stellate cells, while perpetuation is the response to stimuli that maintains the activated stellate cell in its myofibroblastic state to allow fibrotic scar formation.23These changes include stellate cell proliferation, chemotaxis, fibrogenesis, increased contractility, altered matrix degeneration, and cytokine signalling.22
The release of a variety of mitogenic factors causes proliferation of stellate cells. PDGF is a potent mitogen upregulated during liver injury.24Other molecules with mitogenic activity include VEGF, thrombin, epidermal growth factor, TGF-α, keratinocyte growth factor, and fibroblast growth factor.25Matrix metalloproteinase-2 (MMP-2) acts as a stellate cell mitogen via activation by discoidin domain receptor-2 binding to fibrillar collagen.26
Activated stellate cells migrate towards areas of hepatic injury during chemotaxis. Growth factors within the liver which have chemoattractant properties include PDGF, insulin growth factor-1, endothelin-1, monocyte attractant protein-1, and the chemokine receptor CXCR3;27 in contrast, adenosine inhibits chemotaxis, allowing cells to fix at injury sites.28
The primary event of fibrogenesis is increased matrix production and scar formation mediated by stellate cells.10Type I collagen, the major constituent of scar tissue, is upregulated post-transcriptionally in stellate cells by the actions of TGF-αvia Smads, pivotal intracellular effector proteins that mediate TGF-αsignalling.29 CTGF also promotes fibrogenesis.30 Other less potent mediation of Type I collagen production include angiotensin II, IL-1β, and TNF. 31
On activation the HSC contracts, which is characterised by the increased expression of α-smooth muscle actin, a contractile filament protein.32 This results in impendence of sinusoidal blood flow increasing portal resistance, and eventually increasing portal pressure once advanced fibrosis has developed. Progressive development of intrahepatic shunting occurs in conjunction with the constriction of hepatic sinusoids. The process is mediated by endothelin-1, as well as angiotensinogen II and atrial natriuretic peptide. Nitric oxide has an opposing effect, resulting in sinusoidal relaxation.33
The concept of matrix degradation evolved following evidence demonstrating that fibrosis is reversible. This propagated the theory that fibrosis is a dynamic balance between matrix production and degradation.34Initial matrix degradation may be termed pathological, corresponding to disruption of normal low-density matrix of the subendothelial basement membrane, occurring in early stages of fibrosis. This allows for normal matrix to be replaced by a higher density pathological matrix, containing fibrils. Matrix degrading collagenases, including the MMP, break down Type IV collagen and are central to this paradigm.35,36 Stellate cells and Kupffer cells are the primary sources of MMP-2 and MMP-9, respectively.35Conversely, MMP-1 degrades Type I collagen and is involved in the restorative degradation of the pathological matrix, thus, upregulation of this enzyme would favour resolution of scar tissue. Tissue inhibitors of metalloproteinases (TIMP) inhibit the actions of collagenases, resulting in reduced matrix degradation, and inhibit stellate cell apoptosis, which prevents fibrosis resolution and favours matrix accumulation.36TIMP-1 and 2 are upregulated in progressive fibrosis, and their sustained release from stellate cells is a key determinant of progressive fibrosis.36,37
HSC are not only effectors of fibrosis but also have a central role in amplifying inflammatory signaling via toll-like receptors and cytokines including monocyte chemotactic protein-1, CCL21, CCR5, and Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES). They demonstrate antigen presenting capabilities and produce neutrophil chemoattractants.38,39
EXPERIMENTAL MODELS OF LIVER INJURY
Techniques isolating and cultivating HSC have represented a major advance in exploring the complex mechanisms involved in fibrogenesis. Rodent stellate cell lines have been characterised but have now been superseded by human cell lines.23The most utilised human HSC lines include the LX1 and LX2 cell lines. LX1 cell line is generated by transformation with SV40 T antigen.40 The LX2 cell line is generated by isolating stellate cells from normal human livers and immortalising them by culturing in low serum conditions. When activated, these cells bear close similarity to in vivohuman activated HSC.41The LX2 cell line has been extensively validated and used in a number of studies exploring the effects of mediators on stellate cell activity.25 Other human HSC lines are summarised in Table 1.40 Utilising these cell lines therefore allows us the unique opportunity to study factors that both inhibit and activate HSC.
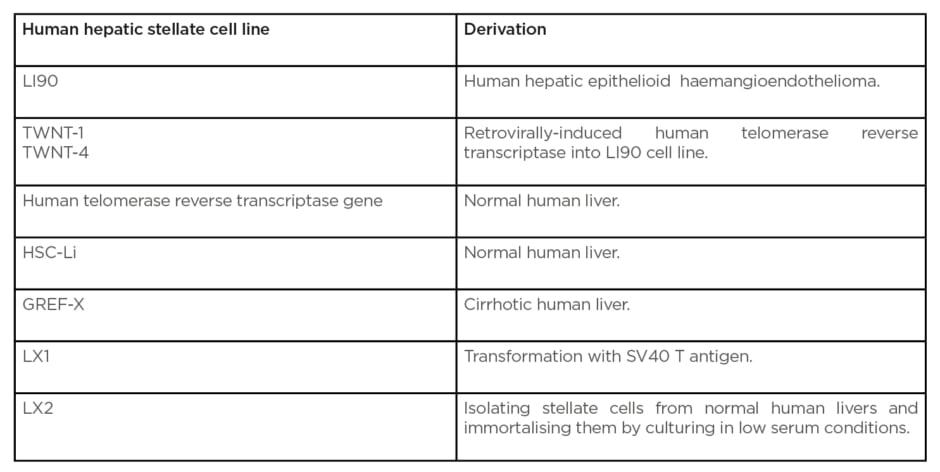
Table 1: Summary table of human stellate cell lines used.
Animal models offer the opportunity to study interactions of different cell types, gene activation, protein-expressing profiles, and signalling pathways, but as yet no animal model exactly reproduces human hepatic fibrosis.42 A variety of agents, often carcinogenic, can be employed to chemically induce fibrosis. The most commonly used are carbon tetrachloride (CCl4) and thioacetamide (TAA). Fibrosis generated in this manner has both a degree of reproducibility and similarity with human mechanisms and pathways involved in hepatic fibrosis.43They also have the added advantage that they can be used in conjunction with either transgenic or wild-type mice to explore underlying molecular mechanisms involved in hepatic fibrosis, or can be used to evaluate the potential of novel antifibrotic agents using strictly controlled environmental and genetic conditions.43
Carbon Tetrachloride
CCL4is the oldest and most common method of inducing liver fibrosis in rodent models.44It is a halothane and induces hepatic injury when given at repetitive low doses. It can be administered in a variety of ways, but is most commonly administered by intraperitoneal injection ≤3 times per week. CCl4is bioactivated by oxidases. This leads to a combination of effects including CCl3radical formation in the liver, which causes hepatocyte damage via lipid peroxidation; HSC activation; Kupffer cell activation; TGF-β-1 upregulation; and increased oxidative stress.45,46Histological alterations result in fatty change with necrosis and intense necroinflammation in centrilobular areas. This progresses to both septal and nonseptal fibrosis resulting in extensive fibrosis and cirrhosis. The fibrous septa are classically thin and can regress with CCl4withdrawal. The reproducibility and ease of induction of fibrosis are its main advantages. Disadvantages include heterogeneity in the amounts of fibrosis produced between animals.32
Thioacetamide
TAA is a selective hepatotoxin. Chronic administration not only induces liver fibrosis, but can result in carcinogenesis, including cholangiocarcinoma and hepatocellular carcinoma. TAA is usually given in drinking water over a period of 8–18 weeks to induce liver fibrosis.47Histologically, mild to moderate amounts of fibrosis develop by 8 weeks with elevated transaminases. By 12 weeks, parenchymal damage occurs, with hepatocyte swelling, necrosis, and proliferation. This results in fibrous enlargement of the portal tracts, with portal–portal and portal–central septa developing resulting in cirrhosis. Histological similarities with human viral hepatitis have led to its use as an indirect model of both fibrosis secondary to HBV and HCV infection. Withdrawal of TAA results in resolution of fibrosis over an 8-week period. A longer period of resolution, compared with CCl4models, makes it a more suitable model when evaluating potential antifibrotic therapies, and the occurrence of regenerative nodules make it more comparable to human cirrhosis.48
Dimethylnitrosamine
Dimethylnitrosamine (DMN) is a hepatotoxin and a carcinogenic and mutagenic agent. DMN is typically administered intraperitoneally three times a week, with centrilobular and periportal fibrosis characteristically developing after 3 weeks. Microscopically, this pattern of fibrosis is seen in cirrhosis.49DMN models induce HSC and Kupffer cells to express profibrotic cytokines resulting in deposition of excessive ECM, the primary pathogenesis of fibrosis, making it a potentially useful animal model.50However, DMN has mutagenic and carcinogenic properties and exposure can cause hepatocellular carcinoma. This makes understanding fibrosis pathways difficult to interpret but does allow the pathogenesis of fibrosis to hepatocellular carcinoma to be better understood.51
BILE DUCT LIGATION
Surgical ligation of the common bile duct causes cholestasis and periportal inflammation. This technique causes proliferation of biliary epithelial cells and increases expression of fibrogenic markers such as TIMP-1, Alpha-SMA, Type I collagen, and TGF-β1.52Although this model aids portal fibrosis and portal myofibroblast interrogation, bile duct ligation model use is restricted due to the high frequency of gall bladder perforation and bilio-peritoneum in mice.53Mortality risk is therefore significant and this model is more suitable for short-term studies investigating cholestasis-induced fibrosis.52,53
ATP-Binding Cassette Subfamily B Member 4
The ATP-binding cassette subfamily B member 4 (Abcb4-/-) gene encodes multidrug resistance 3 (MDR3) protein, the canalicular phosphatidylcholine lipid transporter. Altering this gene by rendering it deficient causes intrahepatic cholestasis and disease similar to that of primary biliary cirrhosis. It functions to protect cellular membranes facing the biliary tree against bile acids and without phosphatidylcholine there is biliary epithelial and ductular damage, portal inflammation and proliferation, and progressive portal fibrosis.54Abcb4-/-knockout mice develop fibrosis between 4 and 8 weeks with TGF-β1 expression, HSC activation at 4 weeks with collagen deposition, and scarring at 8 weeks.55
These models have been used to study primary biliary cirrhosis, cholestasis of pregnancy drug-induced cholestasis, and therapeutic interventions.56
ANTIFIBROTIC THERAPY
Trials of antifibrotic therapies in humans have been limited compared to experimental models in animals. This is partly due to the long duration antifibrotic agents would need to be administered to demonstrate a treatment effect because of the time taken for the agent to systemically accumulate. Trials are limited by funding and concerns over side effects. Furthermore, the historical need for histological endpoints can limit recruitment. An ideal trial would be one that has a short duration of treatment, with a pre-existing clinical need for routine liver histology.
Though significant progress has been made in understanding the pathogenesis of fibrosis, particularly the role of the HSC, no antifibrotic therapy specifically targeting this cell type responsible for increased matrix production and scar formation is currently licensed for human use. Several potential agents and antifibrotic molecules such as silymarin, caffeine, and curcumin, summarised in Table 2, have shown antifibrotic properties via targeting the HSC, but are not routinely used in clinical practice.57,58
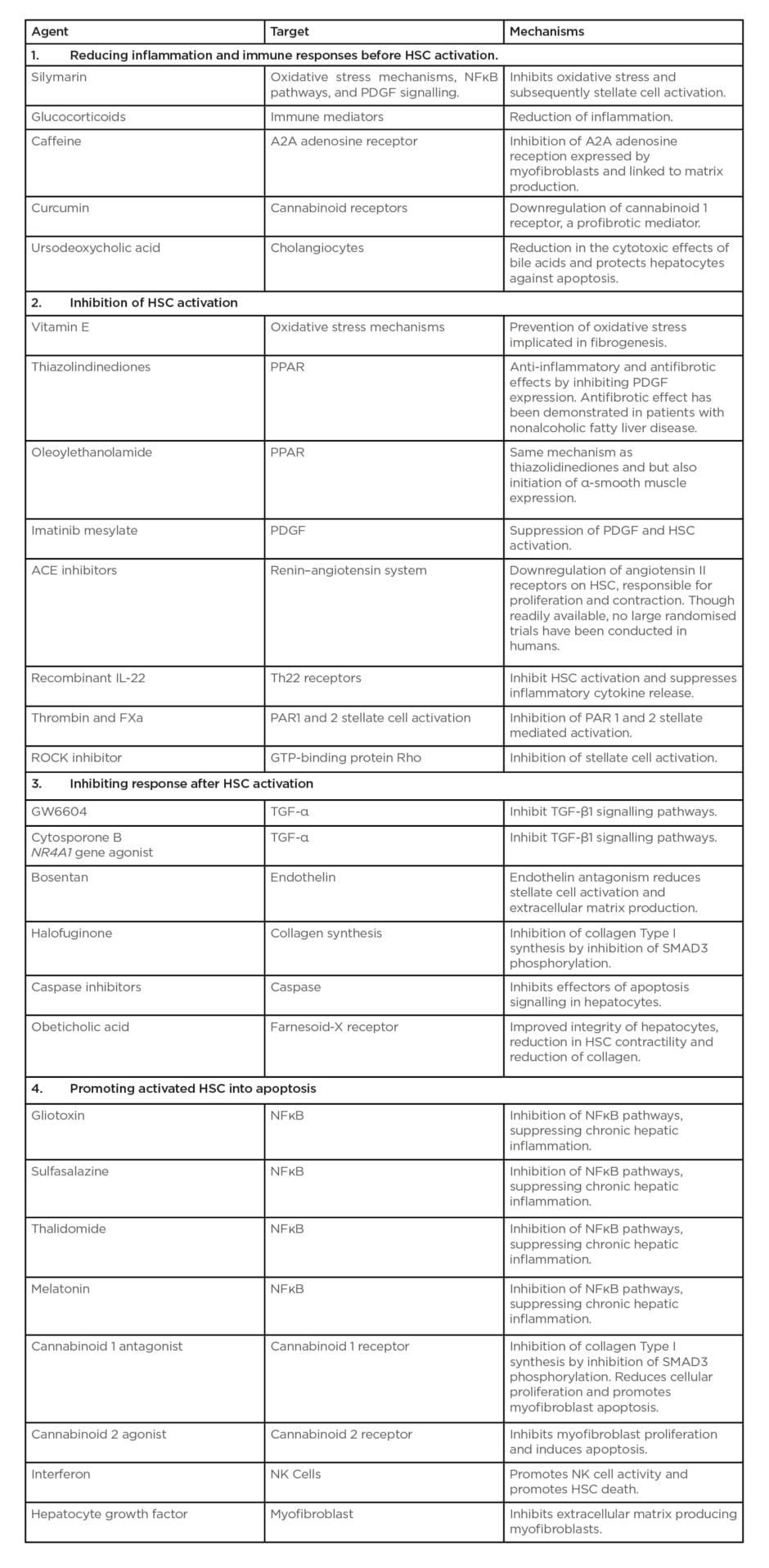
Table 2: Summary table of anti-fibrotic strategies targeting hepatic stellate cell.
ACE: angiotensin-converting enzyme; FXa: Factor Xa; GTP: guanosine-5′-triphosphate; HSC: hepatic stellate cell; NK: natural killer; NR4A1: nuclear receptor subfamily 4 group A member 1; PAR1: protease-activated receptor 1; PAR2: protease-activated receptor 2; PDGF: platelet-derived growth factor; PPAR: peroxisome proliferator activated receptors; ROCK: Rho kinase; SMAD3: SMAD Family Member 3.
Adapted from Ebrahimi H et al.58
Silymarin, mainly consisting of silibinin, is a flavonoid complex extracted from milk thistle. It has been shown to diminish the following: oxidative stress, lysis of hepatocytes, activation of Kupffer cells, and expression of α-SMA and TGF-β1, all implicated in the pathogenesis of fibrosis in rats treated with CCl4to induce fibrosis.59 Despite showing promise in animal models with improved liver function tests, in humans there is little evidence demonstrating its clinical benefit. The De Avelar et al.60meta-analysis of six human studies demonstrated that although alanine transaminase and aspartate transaminase levels were reduced with silymarin use, there was no clinically significant benefit associated with this. To date, only one randomised, double-blinded, placebo-controlled study with biopsy-proven fibrosis investigating the effect of silymarin has been conducted. This showed that silymarin did not significantly reduce nonalcoholic fatty liver disease activity scores, although some improvement of fibrosis was histologically seen compared to placebo. This study was underpowered and did not include other aetiologies of fibrosis.61
Several studies have reported the beneficial effect of caffeine against liver disease.62The major mechanism by which caffeine exerts its antifibrotic effect is largely attributable to caffeine being a pan antagonist of the adenosine receptor. Liver myofibroblasts are profibrogenic and express the A2A adenosine receptor; therefore, blocking this receptor inhibits fibrogenesis.63A second proposed mechanism is that caffeine alters signalling and inflammation pathways in fibrogenesis by reducing TGF-βexpression, as suggested in rodent models of fibrosis.64
Curcumin, a monomer extract from turmeric, has not only shown anti-inflammatory and antiproliferative properties but also antifibrotic actions, as evidenced by its ability to protect against fibrosis in CCl4treated rats.65,66 Curcumin also interferes with TGF-βsignalling pathways and PDGF receptors, leading to inhibition of HSC activity. This is further achieved through modulation of the cannabinoid receptor system with downregulation of the cannabinoid receptor 1, inhibiting ECM expression by HSC.67
Anticoagulation
Evidence suggests that hepatic fibrogenesis is associated with prothrombotic tendencies, including factor V Leiden (FVL); this is demonstrated by Wright et al.,68who found that FVL mutation increased the rate of fibrosis in HCV infection.The coagulation proteins thrombin and factor Xa (FXa) are also implicated through their activation of HSC.69 Activation of the coagulationsystem generates FXa, which in turn results in the production of thrombin from its precursor protein, prothrombin. Both thrombin and FXa activate stellate cells via G-protein-coupled receptors known as protease activated receptors (PAR).70 Duplantier et al.71evaluated, in a rat model of CCl4-induced chronic liver injury, the effect of thrombin inhibition using a synthetic thrombin antagonist SSR182289. The study demonstrated a reduction in liver fibrosis and α-smooth muscle actin expression, a marker of stellate cell activation.Dhar et al.72demonstrated that administration of rivaroxaban, an FXa antagonist, to mice that had been exposed to TAA for 8 weeks induced milder fibrosis, especially around central veins, compared with control mice. The overall mean percentage area of fibrosis was significantly reduced, as was α-smooth muscle actin expression. This effect is likely because of FXa antagonists blocking PAR1 and 2-mediated stellate cell activation.72Prolonged administration of enoxaparin in a rat model of cirrhosis, (induced using CCL4or TAA), resulted in an improvement in both portal hypertension and liver fibrosis, possibly by potentiating fibrosis regression, resulting in reduction of hepatic vascular resistance and portal pressures.73The use of warfarin anticoagulation to reduce liver fibrosis induced by CCl4has been tested using a transgenic mouse model of FVL-activated protein C resistance. The progression of fibrosis in FVL mice was compared to the control C57BL/6 mice which had been anticoagulated with warfarin. The results confirmed that the thrombophilic mouse developed fibrosis at a faster rate than the control mice, but warfarin anticoagulation significantly reduced the rate of fibrosis seen in both strains.74
A small study has suggested an efficacy of low-molecular-weight heparin as an antifibrotic in humans. Patients with HBV infection who were treated with 3 weeks of heparin showed improvedserum levels of alanine aminotransferase and bilirubin, with a reduction in serum hyaluronic acid and Type IV collagen concentrations.75Long-term use of heparin as an antifibrotic agent in humans may not be practical because of side effects including osteopaenia, thrombocytopenia, and idiopathic hepatitis. A multicentre Phase II study evaluated the antifibrotic effect of warfarin anticoagulation in transplanted HCV patients with cirrhosis. Interim results demonstrated a reduction in fibrosis scores at 1-year post-transplantation in warfarinised patients compared to those who were not.76
Angiotensin Converting Enzyme Inhibitors
The role of angiotensin converting enzyme (ACE) inhibitors as antifibrotic agents has been explored with some success. Angiotensinogen and angiotensin-1 are present in hepatocytes and are highly activated in chronic liver disease. Specifically, angiotensin II receptors are upregulated during chronic liver injury resulting in HSC activation. Activation of the renin–angiotensin–aldosterone system also occurs within the liver, triggering oxidative stress and inflammatory cell release contributing to fibrogenesis.77The use of ACE inhibitors in preventing HSC, and renin–angiotensin system activation in preventing fibrogenesis, is a promising treatment option. This has been supported in animal studies, which demonstrated that ACE inhibitor use in CCl4treated rats ameliorated levels of oxidative stress, hepatic inflammation, and hepatic fibrosis.78 A meta-analysis by Kim et al.78confirmed the benefits of their use in reducing hepatic fibrosis in humans. This meta-analysis only included studies in which intervention groups used ACE inhibitors or angiotensin receptor blockers and compared their effect to placebo. Histological changes in fibrosis were the primary outcome. ACE inhibitors lowered fibrosis scores, serum fibrosis markers including TGF-β-1, collagen Types I and IV, TIMP-1, and MMP-2. Significantly, they were shown to be safe with no significant differences in renal function in those who received ACE inhibitors versus those who did not.78
Farnesoid X Receptor Agonists
The potent farnesoid X receptor (FXR) agonist obeticholic acid has been shown to have antifibrotic effects, especially in those with primary biliary cirrhosis and nonalcoholic steatohepatitis, histologically characterised by the presence of fibrosis. FXR is present on HSC and is involved in cellular regulation and activation. FXR agonists, therefore, have the ability to inhibit HSC activation and hepatic fibrogenesis.79Obeticholic acid use in TAA-treated rats decreased hepatic inflammation and fibrogenesis, as well as portal pressure and intrahepatic vascular resistance. There was also decreased profibrotic cytokine activity, assessed by TGF-β-1, CTGF, and PDGF.80 The FLINT trial, a Phase IIb nonalcoholic steatohepatitis study, compared those treated with 72 weeks of obeticholic acid versus placebo and fibrosis improvement was the primary outcome of the study. It assessed for statistically significant improvement in hepatic fibrosis and fibrosis scores observed in the treatment arm compared to those not being treated.79Although the antifibrotic properties of FXR agonists are significant, mild-to-moderate side effects including pruritis, dyslipidaemia, fatigue, headache, and gall stone disease have been reported, which may limit their use in clinical practice.79
Thiazolidinediones
Thiazolidinediones, including pioglitazone and rosiglitazone, are peroxisome proliferator-activated receptors (PPAR) agonists and are routinely used in the treatment of diabetes. They are selective ligands for nuclear transcription factor PPAR, expressed in HSC. PPAR activation downregulates the ability to proliferate and migrate in response to PDGF and reduces expression of TGF-β-1, procollagen 1, fibronectin, and TIMP, inhibiting the fibrogenic process. Thiazolidinediones have been shown to be effective antifibrotic agents in CCl4treated rats, with positive improvements in histology and profibrotic cytokine profiling.81A meta-analysis of 8 randomised clinical trials, including 516 patients diagnosed radiologically or histologically, concluded that thiazolidinedione use for 2 years reversed advanced fibrosis and improved fibrosis stage, even in those without diabetes.82
CONCLUSION
Our understanding of the pathophysiology of liver fibrosis continues to grow. Numerous experimental studies have demonstrated agents that may have therapeutic potential as antifibrotic agents. Despite this, there are still few candidates that have been successfully trialled in human clinical trials that have demonstrated a clear and significant antifibrotic effect. More recently, potential therapies have shown increasing promise and further studies are required to ascertain whether these therapies will be translated to clinical practice. At present, removal of the insult resulting in fibrosis remains a key strategy in its reduction and prevention.